TRABAJO GRUPOS 4,5,6,7, A DE LA TABLA PERIÓDICA
KAROLL LIZETH PAZ HERNANDEZ
INSTITUCIÓN EDUCATIVA EXALUMNAS DE LA PRESENTACIÓN
QUIMICA
ONCE-TRES
2019
GRUPO 4A DE LA TABLA PERIÓDICA
El grupo 4A de la tabla periódica de los elementos, también conocido como grupo 14, grupo del carbono o de los carbonoideos, está formado por los siguientes elementos: carbono (C), silicio (Si), germanio (Ge), estaño (Sn) y plomo (Pb).
La mayoría de los elementos de este grupo son muy conocidos y difundidos, especialmente el carbono, elemento fundamental de la química orgánica. A su vez, el silicio es uno de los elementos más abundantes en la corteza terrestre (28%), y de gran importancia en la sociedad a partir del siglo XX, ya que es el elemento principal de los circuitos integrados.
Al bajar en el grupo, estos elementos van teniendo características cada vez más metálicas: el carbono es un no metal, el silicio y el germanio son semimetales, y el estaño y el plomo son metales.
Los elementos que están en este grupo forman más de la cuarta parte de la corteza terrestre y solo podemos encontrar en forma natural al carbono, al estaño y al plomo en forma de óxidos y sulfuros.
La configuración electrónica de todos los elementos de este grupo termina en ns2,p2, debido a que al estar en el grupo 4 poseen en su capa de valencia el orbital s completo y llegan a 2 electrones en el orbital p ya que son el segundo grupo de dicho bloque (p).

CARBONO
Es un elemento químico de número atómico 6, es un sólido a temperatura ambiente. Es el pilar básico de la química orgánica; se conocen cerca de 16 millones de compuestos de carbono, aumentando este número en unos 500.000 compuestos por año, y forma parte de todos los seres vivos conocidos. Forma el 0,2 % de la corteza terrestre.
El grupo de carbono es un grupo de la tabla periódica integrado por los elementos: carbono (C), silicio (Si), germanio (Ge), estaño (Sn), plomo (Pb) En la notación moderna de la IUPAC se lo llama Grupo 14. En el campo de la física de los semiconductores, todavía es universalmente llamado Grupo IV.

Propiedades químicas
Al igual que otros grupos, los miembros de esta familia poseen similitudes en su configuración electrónica, ya que poseen la misma cantidad de electrones en el último nivel o subnivel de energía. Eso explica las similitudes en sus comportamientos químicos Cada uno de los elementos de este grupo tiene 4 electrones en su capa más externa. En la mayoría de los casos, los elementos comparten sus electrones; la tendencia a perder electrones aumenta a medida que el tamaño del átomo aumenta. El carbono es un no metal que forma iones negativos bajo forma de carburos (4-). El silicio y el germanio son metaloides con número de oxidación +4. El estaño y el plomo son metales que también tienen un estado de oxidación +2. El carbono forma tetrahaluros con los halógenos. El carbono se puede encontrar bajo la forma de tres óxidos: dióxido de carbono (CO2), monóxido de carbono (CO) y dióxido de tricarbono (C3O2).El carbono forma disulfuros y diselenios.1
| Z | Elemento | Distribución electrónica/valencia |
|---|---|---|
| 6 | Carbono | 2, 4 |
El silicio forma dos hidruros: SiH4 y Si2H6. El silicio forma tetrahaluros de silicio con flúor, cloro e yodo. El silicio también forma un dióxido y un disulfuro.La fórmula química del nitruro de silicio es Si3N4.2
El germanio forma dos hidruros: GeH4 y Ge2H6. El germanio también fomrma tetrahaluros con todos los halógenos, excepto con el astato y forma di dihaluros con todos los halógenos excepto con el bromo y el astato. El Germanio también forma dióxidos, disulfuros y diselenios.
El estaño forma dos hidruros: SnH4 y Sn2H6. El estaño forma tetrahaluros y dihaluros con todos los halógenos menos con el Astato.
El plomo forma hidruros bajo la forma de PbH4. Forma dihaluros y tetrahaluros con el flúor y con el cloro. También forma tetrabromuros y dihioduros.

Propiedades físicas
Los puntos de ebullición en el grupo del carbono tienden a disminuir a medida que se desciende en el grupo. El carbono es el más ligero del grupo, el mismo sublima a 3825°C.El punto de ebullición del silicio es 3265°C, el del germanio es 2833°C, el del estaño es 2602°C y el del plomo es 1749°C. Los puntos de fusión tienen la misma tendencia que su punto de ebullición. El punto de fusión del silicio es 1414°C, el del germanio 939°C, para el estaño es 232°C y para el plomo 328°C.3
La estructura cristalina del carbono es hexagonal, a altas presiones y temperaturas se encuentra bajo la forma de diamante.
La densidad de los elementos del grupo del carbono tiende a aumentar con el aumento del número atómico. El carbono tiene una densidad de 2,26 g/cm3, la densidad del silicio es de 2,33 g/cm3 y la densidad del germanio es de 5,32 g/cm3. El estaño tiene una densidad de 7,26 g/cm3 mientras que la del plomo es de 11,3 g/cm
El radio atómico de los elementos del grupo del carbono tiende a aumentar a medida que aumenta el número atómico. El radio atómico del carbono es de 77 picometros, el del silicio es de 118 picómetros, el del germanio es de 123 picómetros, el del estaño es de 141 picómetros, mientras que el del plomo es de 175 picómetros.
Alótropos
El carbono posee varios alótropos. El más común es el grafito, que es el carbono en forma de hojas apiladas. Otra forma de carbono es el diamante. Una tercera forma alotrópica del carbono es el fullereno, que tiene la forma de láminas de átomos de carbono dobladas que forman una esfera. Un cuarto alótropo del carbono, descubierto en 2003, se llama grafeno, y está en forma de una capa de átomos de carbono dispuestos en forma similar a la de un panal.45
El silicio tiene dos alótropos, el amorfo y el cristalino. El alótropo amorfo es un polvo marrón, mientras que el alótropo cristalino es gris y tiene un brillo metálico.6
El estaño tiene dos alótropos: α-estaño, también conocido como estaño gris, y β-estaño. El estaño se encuentra típicamente en la forma β-estaño. Sin embargo a presión normal el β-estaño se convierte a α-estaño, pasando de un metal plateado a un polvo gris, a temperaturas inferiores a los 56º Fahrenheit. Esto puede hacer que los objetos de estaño a temperaturas bajas se desmoronen en un proceso conocido como "la pudrición del estaño".
Núcleo atómico
Al menos dos de los elementos del grupo IV (estaño y plomo) tienen núcleo mágicos, lo que significa que estos elementos son más comunes y más estables que los elementos metálicos que no tiene un núcleo mágico.
Isótopos
Existen 15 isótopos conocidos de carbono. De ellos, tres son de origen natural. El más común de todos ellos es el carbono-12 estable, seguido por el carbono-13 estable.3 El carbono-14 es un isótopo radiactivo natural con una vida media de 5.730 años.
Se han descubierto 23 isótopos de silicio, cinco de ellos son de origen natural. El más común es de silicio-28 estable, seguido de silicio-29 estable y estable de silicio-30. Silicio-32 es un isótopo radiactivo que se produce naturalmente como un resultado de la desintegración radiactiva de los actínidos. Silicio-34 también se produce de forma natural como resultado de la desintegración radiactiva de los actínidos.
Hasta el momento se han descubierto 32 isótopos de Germanio, cinco de ellos son de origen natural. El más común es el isótopo estable de germanio-74, seguido por el isótopo estable de germanio-72, el isótopo estable de germanio-70, y el isótopo estable de germanio-73. El isótopo de germanio-76 es un radioisótopo.
Se han descubierto 40 isótopos de estaño, 14 de ellos se producen en la naturaleza. El más común es el isótopo estable estaño-120, seguido por el isótopo estable estaño-118, el isótopo estable estaño-116, el isótopo estable estaño-119, el isótopo estable estaño-117, el radioisótopo estaño-124, el isótopo estable estaño-122m el isótopo estable estaño-112 y el isótopo estable estaño-114. El estaño también tiene cuatro radioisótopos que se producen como resultado de la desintegración radiactiva de uranio. Estos isótopos son el estaño-121, estaño-123, estaño-125, y el estaño-126.
Se han descubierto 38 isótopos de plomo, 9 de ellos son de origen natural. El isótopo más común es el radioisótopo plomo-208, seguido por el plomo-206, el radioisótopo plomo-207, y el radioisótopo plomo-204. Cuatro isótopos de plomo se producen a partir de la desintegración radiactiva del uranio y el torio. Estos isótopos son el plomo-209, el plomo-210, el plomo-211 y plomo-212.

Descubrimiento y usos en la antigüeda
El carbono, estaño y plomo son algunos de los elementos bien conocidos en el mundo antiguo, junto con azufre, hierro, cobre, mercurio, plata y oro.7
Carbono como elemento fue utilizado por el primer ser humano para manejar carbón de un incendio.
El Silicio como sílice en forma de cristal de roca era familiar a los egipcios predinásticos, que lo utilizaron para los granos y pequeños jarrones. La fabricación de vidrio que contiene sílice se llevó a cabo tanto por los egipcios - al menos desde 1500 A.C - y por los fenicios. Muchos de los compuestos de origen natural o minerales de silicato fueron utilizados en diversos tipos de mortero para la construcción de viviendas.
Los orígenes de estaño parecen estar perdido en la historia. Parece que el bronce, que es una aleación de cobre y estaño, fue utilizado por el hombre prehistórico y algún tiempo antes se aisló el metal puro. Minas de estaño operaban tanto en la zonas aztecas de Sur y Centro América Inca y antes de la conquista española.
El plomo se menciona a menudo en relatos bíblicos. Los babilonios utilizaban el metal en forma de placas en los que grababan inscripciones. Los romanos lo utilizaron para las tabletas, tuberías de agua, monedas y utensilios de cocina; de hecho, como resultado de la última utilización, el envenenamiento por plomo fue reconocido en la época de Augusto César. El compuesto conocido como blanco de plomo aparentemente se preparó como un pigmento decorativo al menos desde 200 aC.
Aplicaciones
El carbono es comúnmente utilizado en su forma amorfa. En esta forma el carbono se utiliza para la fabricación de acero, como relleno en los neumáticos, y como carbón activado. El carbono grafito se utiliza en los lápices. El diamante, otra de las formas del carbono, se utiliza comúnmente en la joyería. Las fibras de carbono se utilizan en numerosas aplicaciones, tales como puntales de satélite, debido a que las fibras son muy fuertes pero elásticas.8
El dióxido de silicio tiene una amplia variedad de aplicaciones, incluyendo pasta de dientes,materiales de construcción, y la sílice es un uno de los componentes principales del vidrio. Un 50% del silicio puro se dedica a la fabricación de aleaciones de metales. Mientras que un 45% se dedica a la fabricación de siliconas. El silicio también se usa comúnmente en los semiconductores desde la década de 1950.
El germanio se utilizó en los semiconductores hasta la década de 1950, cuando fue sustituido por el silicio. Los detectores de radiación contienen germanio. El óxido de germanio se utiliza en la fibra óptica.
El uso más importante del estaño es en soldaduras; 50% de todo el estaño producido se destina a esta aplicación. Un 20% del estaño producido se utiliza en la hojalata. Otro 20% del estaño se utiliza en la industria química. El óxido de estaño (IV) se utiliza comúnmente en la cerámica desde hace miles de años.
Alguna de las aplicaciones del plomo son las pesas, pigmentos y como protección contra materiales radioactivos. El plomo fue utilizado históricamente en la gasolina en forma de tetraetilo de plomo, pero este uso se ha interrumpido debido a su alta toxicidad.

SILICIO
El silicio (del latín: sílex) es un elemento químico metaloide, número atómico 14 y situado en el grupo 14 de la tabla periódica de los elementos de símbolo Si.1 Es el segundo elemento más abundante en la corteza terrestre (25,7 % en peso)2 después del oxígeno. Se presenta en forma amorfa y cristalizada; el primero es un polvo parduzco, más activo que la variante cristalina, que se presenta en octaedros de color azul grisáceo y brillo metálico.

Características



Sus propiedades son intermedias entre las del carbono y el germanio. En forma cristalina es muy duro y poco soluble y presenta un brillo metálico y color grisáceo.3 Aunque es un elemento relativamente inerte y resiste la acción de la mayoría de los ácidos, reacciona con los halógenos4 y álcalis diluidos. El silicio transmite más del 95 % de las longitudes de onda de la radiación infrarroja.
Se prepara en forma de polvo amarillo pardo o de cristales negros-grisáceos. Se obtiene calentando sílice, o dióxido de silicio (SiO2), con un agente reductor, como carbono o magnesio, en un horno eléctrico.5 El silicio cristalino tiene una dureza de 7, suficiente para rayar el vidrio, de dureza de 5 a 7. El silicio tiene un punto de fusión de 1.411 °C, un punto de ebullición de 2.355 °C y una densidadrelativa de 2,33(g/ml). Su masa atómica es 28,086 u (unidad de masa atómica).
Se disuelve en ácido fluorhídrico formando el gas tetrafluoruro de silicio, SiF4(ver flúor), y es atacado por los ácidos nítrico, clorhídrico y sulfúrico, aunque el dióxido de silicio formado inhibe la reacción. También se disuelve en hidróxido de sodio, formando silicato de sodio y gas hidrógeno. A temperaturas ordinarias el silicio no es atacado por el aire, pero a temperaturas elevadas reacciona con el oxígeno formando una capa de sílice que impide que continúe la reacción. A altas temperaturas reacciona también con nitrógeno y cloro formando nitruro de silicio y cloruro de silicio, respectivamente.
El silicio constituye un 28 % de la corteza terrestre. No existe en estado libre, sino que se encuentra en forma de dióxido de silicio y de silicatos complejos. Los minerales que contienen silicio constituyen cerca del 40 % de todos los minerales comunes, incluyendo más del 90 % de los minerales que forman rocas volcánicas. El mineral cuarzo, sus variedades (cornalina, crisoprasa, ónice, pedernal y jaspe) y los minerales cristobalita y tridimita son las formas cristalinas del silicio existentes en la naturaleza. El dióxido de silicio es el componente principal de la arena. Los silicatos (en concreto los de aluminio, calcio y magnesio) son los componentes principales de las arcillas, el suelo y las rocas, en forma de feldespatos, anfíboles, piroxenos, micas y zeolitas, y de piedras semipreciosas como el olivino, granate, zircón, topacio y turmalina.
Silicio como base bioquímica[editar]
Sus características compartidas con el carbono, como estar en la misma familia 14, no ser metal propiamente dicho, poder construir compuestos parecidos a las enzimas (zeolitas), otros compuestos largos con oxígeno (siliconas) y poseer los mismos cuatro enlaces básicos, le confiere cierta oportunidad en llegar a ser base de seres vivos, aunque no sea en la Tierra, en una bioquímica hipotética.
Aplicaciones
Se utiliza en aleaciones, en la preparación de las siliconas, en la industria de la cerámica técnica y, debido a que es un material semiconductor muy abundante, tiene un interés especial en la industria electrónica y microelectrónica como material básico para la creación de obleas o chips que se pueden implantar en transistores, pilas solares y una gran variedad de circuitos electrónicos. El silicio es un elemento vital en numerosas industrias. El dióxido de silicio (arena y arcilla) es un importante constituyente del hormigón y los ladrillos, y se emplea en la producción de cemento portland. Por sus propiedades semiconductoras se usa en la fabricación de transistores, células solares y todo tipo de dispositivos semiconductores; por esta razón se conoce como el Valle del Silicio a la región de California en la que concentran numerosas empresas del sector de la electrónica y la informática. También se están estudiando las posibles aplicaciones del siliceno, que es una forma alotrópica del silicio que forma una red bidimensional similar al grafeno. Otros importantes usos del silicio son:
- Como material refractario, se usa en cerámicas, vidriados y esmaltados.
- Como elemento fertilizante en forma de mineral primario rico en silicio, para la agricultura.
- Como elemento de aleación en fundiciones.
- Fabricación de vidrio para ventanas y aislantes.
- El carburo de silicio es uno de los abrasivos más importantes.
- Se usa en láseres para obtener una luz con una longitud de onda de 456 nm.
- La silicona se usa en medicina en implantes de seno y lentes de contacto.
Se utiliza en la industria del acero como componente de las aleaciones de silicio-acero. Para fabricar el acero, se desoxida el acero fundido añadiéndole pequeñas cantidades de silicio; el acero común contiene menos de un 0,30 % de silicio. El acero al silicio, que contiene de 2,5 a 4 % de silicio, se usa para fabricar los núcleos de los transformadores eléctricos, pues la aleación presenta baja histéresis (véase Magnetismo). Existe una aleación de acero, el durirón, que contiene un 15 % de silicio y es dura, frágil y resistente a la corrosión; el durirón se usa en los equipos industriales que están en contacto con productos químicos corrosivos. El silicio se utiliza también en las aleaciones de cobre, como el bronce y el latón.
Abundancia y obtención[editar]
El silicio es uno de los componentes principales de los aerolitos, una clase de meteoroides.
Medido en peso, el silicio representa más de la cuarta parte de la corteza terrestre y es el segundo elemento más abundante por detrás del oxígeno. El silicio no se encuentra en estado nativo; arena, cuarzo, amatista, ágata, pedernal, ópalo y jaspe son algunos de los minerales en los que aparece el óxido, mientras que formando silicatos se encuentra, entre otros, en el granito, feldespato, arcilla, hornblenda y mica.
Los métodos físicos de purificación del silicio metalúrgico[editar]
Estos métodos se basan en la mayor solubilidad de las impurezas en el silicio líquido, de forma que éste se concentra en las últimas zonas solidificadas. El primer método, usado de forma limitada para construir componentes de radar durante la Segunda Guerra Mundial, consiste en moler el silicio de forma que las impurezas se acumulen en las superficies de los granos; disolviendo éstos parcialmente con ácido se obtenía un polvo más puro. La fusión por zonas, el primer método usado a escala industrial, consiste en fundir un extremo de la barra de silicio y trasladar lentamente el foco de calor a lo largo de la barra de modo que el silicio va solidificando con una pureza mayor al arrastrar la zona fundida gran parte de las impurezas. El proceso puede repetirse las veces que sea necesario hasta lograr la pureza deseada bastando entonces cortar el extremo final en el que se han acumulado las impurezas.
Los métodos químicos de purificación del silicio metalúrgico[editar]

Los métodos químicos, usados actualmente, actúan sobre un compuesto de silicio que sea más fácil de purificar descomponiéndolo tras la purificación para obtener el silicio. Los compuestos comúnmente usados son el triclorosilano (HSiCl3), el tetracloruro de silicio (SiCl4) y el silano (SiH4).
En el proceso Siemens,6 las barras de silicio de alta pureza se exponen a 1150 °C al triclorosilano, gas que se descompone depositando silicio adicional en la barra según la siguiente reacción:
- 2 HSiCl3 → Si + 2 HCl + SiCl4
El silicio producido por éste y otros métodos similares se denomina silicio policristalino y típicamente tiene una fracción de impurezas de 0,001 ppm o menor.
El método Dupont consiste en hacer reaccionar tetracloruro de silicio a 950 °C con vapores de cinc muy puros:
- SiCl4 + 2 Zn → Si + 2 ZnCl2
Este método está plagado de dificultades (el cloruro de cinc, sub producto de la reacción, solidifica y obstruye las líneas), por lo que eventualmente se ha abandonado en favor del proceso Siemens.
Una vez obtenido el silicio ultrapuro es necesario obtener un monocristal, para lo que se utiliza el proceso Czochralski.
Alternativas metalúrgicas[editar]
También se han realizado grandes esfuerzos en conseguir SoG-Si evitando el paso energéticamente costoso del uso de triclorosilano, silano o tetraclorosilano, y el posterior depósito en Siemens o similares.
Elkem11 purifica mg-Si en tres pasos de refino relativamente simples, pirometalúrgico, hidrometalúrgico, y de limpieza, con un consumo de sólo el 20 al 25 % de la energía utilizada en la ruta Siemens. Junto con la Universidad de Constanza, han conseguido eficiencias de célula sólo medio punto por debajo de las células comerciales.
Apollon Solar SAS y el laboratorio nacional de investigación francés CNRS purifican Mg-Si con un plasma. Se han conseguido células solares de un 11,7 % de eficiencia.12
Otra alternativa metalúrgica es producir mg-Si con cuarzo y carbón negro tan puros que no sea necesario refinarlo más. Hay dos trabajos en paralelo: uno es el de la Universidad Nacional Técnica de Kazakh en Almaty, Kazajistán.7 El otro es el proyecto SOLSILC, financiado por la Comisión Europea. Las células solares fabricadas con este material han obtenido eficiencias de momento relativamente bajas.8 28 por ciento de este material ya no existe.
Isótopos[editar]
El silicio tiene nueve isótopos, con número másico entre 25 a 33. El isótopo más abundante es el Si-28 con una abundancia del 92,23 %, el Si-29 tiene una abundancia del 4,67 % y el Si-30 que tiene una abundancia del 3,1 %. Todos ellos son estables teniendo el resto de isótopos una proporción ínfima. El Si-32 es un isótopo radiactivo que proviene del decaimiento del argón. Su tiempo de semivida es aproximadamente de unos 132 años. Padece un decaimiento beta que lo transforma en P-32 (que tiene un periodo de semivida de 14,28 días).

GERMANIO
El germanio (antiguamente llamado eka-silicio) es un elemento químico con número atómico 32, y símbolo Geperteneciente al período 4 de la tabla periódica de los elementos.
Características principales[editar]

Es un semimetal, de color blanco grisáceo lustroso, quebradizo, que conserva el brillo a temperaturas ordinarias. Presenta la misma estructura cristalina que el diamante y resiste a los ácidos y álcalis.
Forma gran número de compuestos organometálicos y es un importante material semiconductor utilizado en transistores y fotodetectores. A diferencia de la mayoría de semiconductores, el germanio tiene una pequeña banda prohibida (band gap) por lo que responde de forma eficaz a la radiación infrarroja y puede usarse en amplificadores de baja intensidad.
Aplicaciones[editar]
Las aplicaciones del germanio se ven limitadas por su elevado costo y en muchos casos se investiga su sustitución por materiales más económicos.
- Fibra óptica.
- Electrónica: radares y amplificadores de guitarras eléctricas usados para recrear sonidos de la primera época del rock and roll; aleaciones de Germanato de Silicio (SiGe) en circuitos integrados de alta velocidad. También se utilizan compuestos sandwich Si/Ge para aumentar la movilidad de los electrones en el silicio (streched silicon).
- Óptica de infrarrojos: Espectroscopios, sistemas de visión nocturna y otros equipos.
- Lentes, con alto índice de refracción, de ángulo ancho y para microscopios.
- En joyería se usa la aleación Au con 12% de germanio.
- Como elemento endurecedor del aluminio, magnesio y estaño.
- Quimioterapia.
- El tetracloruro de germanio es un ácido de Lewis y se usa como catalizador en la síntesis de polímeros (PET).
Historia[editar]
Las propiedades del germanio (del latín Germania, Alemania) fueron predichas en 1871 por Mendeleyev en función de su posición en la tabla periódica, elemento al que llamó eka-silicio. El alemán Clemens Winkler demostró en 1886 la existencia de este elemento, descubrimiento que sirvió para confirmar la validez de la tabla periódica, habida cuenta de las similitudes entre las propiedades predichas y las observadas:
| Propiedad | Ekasilicio | Germanio |
|---|---|---|
| (Predichas, 1871) | (Observadas, 1886) | |
| Masa atómica | 72 | 72,59 |
| Densidad (g/cm3) | 5,5 | 5,35 |
| Calor específico (kJ/kg·K) | 0,31 | 0,32 |
| Punto de fusión (°C) | alto | 960 |
| Fórmula del óxido | RO2 | GeO2 |
| Fórmula del cloruro | RCl4 | GeCl4 |
| Densidad del óxido (g/cm3) | 4,7 | 4,7 |
| Punto de ebullición del cloruro (°C) | 100 | 86 |
| Color | gris | gris |
Abundancia y obtención[editar]
Se obtiene de yacimientos de plata, zinc y cobre. Los únicos minerales rentables para la extracción del germanio son la germanita (69% de Ge) y garnierita (7-8% de Ge); además está presente en el carbón, la argirodita y otros minerales. La mayor cantidad, en forma de óxido (GeO2), se obtiene como subproducto de la obtención del zinc o de procesos de combustión de carbón (en Rusia y China se encuentra el proceso en desarrollo).
La purificación del germanio pasa por su tetracloruro que puede ser destilado y luego es reducido al elemento con hidrógeno o con magnesio elemental.
Con pureza del 99,99%, para usos electrónicos se obtiene por refino mediante fusión por zonas resultando cristales de 25 a 35 mm usados en transistores y diodos; con esta técnica las impurezas se pueden reducir hasta 0,0001 ppm.
El desarrollo de los transistores de germanio abrió la puerta a numerosas aplicaciones electrónicas que hoy son cotidianas. Entre 1950 y a principios de los 70, la electrónica constituyó el grueso de la creciente demanda de germanio hasta que empezó a sustituirse por el silicio por sus superiores propiedades eléctricas. Actualmente la gran parte del consumo se destina a fibra óptica (cerca de la mitad), equipos de visión nocturna y catálisis en la polimerización de plásticos, aunque se investiga su sustitución por catalizadores más económicos. En el futuro es posible que se extiendan las aplicaciones electrónicas de las aleaciones silicio-germanio en sustitución del arseniuro de galio especialmente en las telecomunicaciones sin cable.
Isótopos[editar]
El germanio tiene cinco isótopos estables siendo el más abundante el Ge-74 (35,94%). Se han caracterizado 18 radioisótopos de germanio, siendo el Ge-68 el de mayor vida media con 270,8 días. Se conocen además 9 estados metaestables.
Precauciones[editar]
Algunos compuestos de germanio (tetrahidruro de germanio o germano) tienen una cierta toxicidad en los mamíferos pero son letales para algunas bacterias. También es letal para la taenia.
Toxicidad[editar]
El germanio se encuentra más comúnmente en la naturaleza como un contaminante de diversos minerales y es obtenido de los residuos de cadmio remanentes del procesado de los minerales de zinc. Las investigaciones toxicológicas han demostrado que el germanio no se localiza en ningún tejido dado que se excreta rápidamente principalmente por la orina. Las dosis excesivas de germanio lesionan los lechos capilares de los pulmones. Produce una diarrea muy marcada que provoca una deshidratación, hemoconcentración, caída de la presión arterial e hipotermia.

ESTAÑO
El estaño es un elemento químico de símbolo Sn (del latín stannum) y número atómico 50. Está situado en el grupo 14 de la tabla periódica de los elementos. Se conocen 10 isótopos estables. Su principal mena es la casiterita.
Características del estaño[editar]

Es un metal blanco, maleable, que se oxida fácilmente, a temperatura ambiente, cambiando de color a un gris más opaco, y es resistente a la corrosión. Se encuentra en muchas aleaciones y se usa para recubrir otros metales protegiéndolos de la corrosión. Al doblar una barra de este metal se produce un sonido característico llamado grito del estaño, producido por la fricción de los cristales que la componen. Una de sus características más llamativas es que bajo determinadas condiciones sufre la peste del estaño. Por debajo de los -18°C empieza a descomponerse y a convertirse en un polvo gris; a este proceso se lo conoce como peste del estaño. El estaño puro tiene dos variantes alotrópicas: el estaño gris, polvo no metálico, semiconductor, de estructura cúbica y estable a temperaturas inferiores a 13,2 °C, que es muy frágil y tiene un peso específico más bajo que el blanco. El estaño blanco, el normal, metálico, conductor eléctrico, de estructura tetragonal y estable a temperaturas por encima de 13,2 °C.
Usos[editar]
- Se usa como protector del oro, del acero y de diversos metales usados en la fabricación de latas de conserva.
- También se usa para disminuir la fragilidad del vidrio.
- Los compuestos de estaño se usan para fungicidas, tintes, dentífricos y pigmentos.
- Se usa para realizar bronce, aleación de estaño y cobre.
- Se usa para la soldadura blanda, aleado con plomo.
- Se usa en aleación con plomo para fabricar la lámina de los tubos de los órganos musicales.
- Tiene utilidad en etiquetas.
- Recubrimiento de acero.
- Se usa como material de aporte en soldadura blanda con cautín, bien puro o aleado. La directiva RoHS prohíbe el uso de plomo en la soldadura de determinados aparatos eléctricos y electrónicos.
- El estaño también se utiliza en la industria de la cerámica para la fabricación de los esmaltes cerámicos. Su función es la siguiente: en baja y en alta es un opacificante. En alta la proporción del porcentaje es más alto que en baja temperatura.
- Es usado también en el sobretaponado de botellas de vino, en forma de cápsula. Su uso se extendió tras la prohibición del uso del plomo en la industria alimentaria. España es uno de los mayores fabricantes de cápsulas de estaño.
Efectos toxicológicos[editar]
Tanto el estaño metálico como sus compuestos orgánicos e inorgánicos, ya sean formados de manera natural o en sus usos industriales, puede producir efectos tóxicos sobre el medio ambiente y los seres vivos expuestos a ellos.
El estaño es liberado en el medio ambiente por procesos naturales y por las actividades humanas, tales como la minería, la combustión de petróleo y carbón, además de las actividades industriales asociadas a la producción y usos del estaño.
El estaño metálico cuando se encuentra en la atmósfera en forma gaseosa se adhiere a las partículas de polvo, las cuales pueden ser movilizadas por la acción del viento la lluvia o la nieve.
Cuando se libera el estaño metálico en el medio ambiente, este se puede unir con el cloro, azufre u oxígeno para formar compuestos inorgánicos de estaño, tales como el cloruro de estaño, sulfuro de estaño, u dióxido de estaño. Este tipo de compuestos no pueden ser degradados y solo pueden cambiar su forma química, de manera que son adheridos por el suelo y los sedimentos o son disueltos en el agua.
Cuando se combina con el carbono puede formar compuestos orgánicos tales como dibutilestaño, tributilo de estaño y el trifenilestaño. Este tipo de compuestos pueden ser acumulados en el suelo o en el agua, o ser degradados a compuestos inorgánicos por la acción de la luz solar o las bacterias. El tiempo de permanencia en el medio de estos compuestos es variable en función del compuesto, pudiendo ser desde días hasta meses en el agua, y años si se encuentran en el suelo. Debido a su forma química los compuestos orgánicos de estaño también pueden bioacumularse al ser asimilado por el metabolismo de los seres vivos, sufriendo un proceso de biomagnificación a lo largo de las diferentes redes tróficas.
Efectos sobre el ser humano[editar]
Las principales vías de intoxicación con estaño en humanos son:
- La ingestión de alimentos o bebidas que se encuentran envasados en latas hechas con estaño, aunque la mayoría de las que se encuentran actualmente en el mercado están protegidas mediante una laca protectora.
- Ingestión de pescados o mariscos que procedan de aguas contaminadas con este metal.
- Contacto con productos domésticos que contengan compuestos de estaño, como algunos plásticos tales como el PVC.
- Respirar aire que contenga vapores de estaño o polvo de estaño.
El estaño metálico en sí no es muy tóxico para el ser humano ya que en el tracto digestivo no se absorbe de manera efectiva, pero la inhalación de los vapores de estaño sí que es nociva para el aparato respiratorio.
La ingestión de grandes cantidades de compuestos inorgánicos de estaño puede producir dolores de estómago, anemia, y alteraciones en el hígado y los riñones.
La inhalación o la ingesta de compuestos orgánicos de estaño (tales como el trimetilestaño y el trietilestaño) pueden interferir con el funcionamiento del sistema nervioso y el cerebro. En casos graves, puede causar la muerte. Otros compuestos orgánicos de estaño (tales como el dibutilestaño y el tributilestaño) afectan el sistema inmunitario y a la reproducción en animales, aunque esto no se ha evaluado aún en seres humanos.
Tanto compuestos orgánicos como inorgánicos pueden producir irritación por contacto con la piel o los ojos.

PLOMO
El plomo es un elemento químico de la tabla periódica, cuyo símbolo es Pb (del latín plumbum) y su número atómico es 82 según la tabla actual, ya que no formaba parte en la tabla periódica de Mendeleiev. Este químico no lo reconocía como un elemento metálico común por su gran elasticidad molecular. Cabe destacar que la elasticidad de este elemento depende de la temperatura ambiente, la cual extiende sus átomos.
El plomo es un metal pesado de densidad relativa o gravedad específica 11,4 a 16 °C, de color plateado con tono azulado, que se empaña para adquirir un color gris mate. Es flexible, inelástico y se funde con facilidad. Su fusión se produce a 327,4 °C y hierve a 1725 °C. Las valencias químicas normales son 2 y 4. Es relativamente resistente al ataque del ácido sulfúrico y del ácido clorhídrico, aunque se disuelve con lentitud en ácido nítrico y ante la presencia de bases nitrogenadas. El plomo es anfótero, ya que forma sales de plomo de los ácidos, así como sales metálicas del ácido plúmbico. Tiene la capacidad de formar muchas sales, óxidos y compuestos organometálicos.

Características generales[editar]
Los compuestos de plomo más utilizados en la industria son los óxidos de plomo, el tetraetilo de plomo y los silicatos de plomo. El plomo forma aleaciones con muchos metales, y, en general, se emplea en esta forma en la mayor parte de sus aplicaciones. Es un metal pesado y tóxico, y la intoxicación por plomo se denomina como saturnismo o plumbosis.
Isótopos del plomo[editar]
El plomo está constituido por muchos isótopos, siendo estables cuatro de ellos: 204Pb, 206Pb, 207Pb, y 208Pb.
Al 204Pb se le conoce como plomo primordial, y el 206Pb, 207Pb y 208Pb se forman por la desintegración radioactiva de dos isótopos del uranio (235U y 238U) y un isótopo del torio (232Th).
El 210Pb es radioactivo y un precursor del 210Po en la serie de decaimiento del 238U.
La concentración de 210Pb en fumadores es el doble que la concentración en no fumadores. Esta diferencia se atribuye a la inhalación de 210Pb en el humo del tabaco.12
Fuentes de plomo[editar]

El plomo rara vez se encuentra en su estado elemental. Se presenta comúnmente como sulfuro de plomo en la galena.3 Otros minerales de importancia comercial son los carbonatos (cerusita, PbCO3)3 y los sulfatos(anglesita, PbSO4).3 Los fosfatos (piromorfita, Pb5Cl(PO4)3),3 los vanadatos (vanadinita, Pb5Cl(VO4)3),3 los arseniatos (mimelita, Pb5Cl(AsO4)3),3 los cromatos (crocoita, PbCrO4)3 y los molibdatos(vulferita, PbMoO4),3 los wolframatos (stolzita, PbWO4)3 son mucho menos abundantes. También se encuentra plomo en varios minerales de uranio y de torio, ya que proviene directamente de la desintegración radiactiva (decaimiento radiactivo).
La mayoría de los minerales contienen menos del 10 % de plomo, y los minerales que contienen tan poco como 3 % de plomo pueden ser explotados económicamente. Los minerales se trituran y se concentran por flotación por espuma típicamente hasta el 70 % o más. Los minerales constituidos por sulfuros se tuestan, produciendo óxido de plomo y principalmente una mezcla de sulfatos y silicatos de plomo y otros metales contenidos en la mena.4 El óxido de plomo del proceso de tostado se reduce en coquede alto horno para obtener el metal.5 En el proceso se separan capas adicionales separados que flotan en la parte superior de la capa de plomo metálico fundido. Estas son escoria (silicatos que contienen 1,5 % de plomo), mate (sulfuros que contienen 15 % de plomo), y speiss (arseniuros de hierro y cobre). Estos residuos contienen concentraciones de cobre, zinc, cadmio y bismuto que pueden ser recuperados económicamente, como puede ser su contenido en plomo sin reducir.4
El plomo metálico que resulta de los procesos de horno de calcinación y alto horno todavía contiene significativas cantidades de contaminantes: arsénico, antimonio, bismuto, zinc, cobre, plata y oro. La masa fundida se trata en un horno de reverbero con aire, vapor y azufre, que oxida los contaminantes excepto plata, oro y bismuto. Los contaminantes oxidados son eliminados como escoria, que flota en la superficie y se retira.46 Dado que las menas de plomo contienen concentraciones significativas de plata, el metal fundido también está generalmente contaminado con plata. La plata metálica, así como el oro se extraen y se recuperan económicamente por medio del proceso Parkes.467 El plomo desplatado se libera del bismuto de acuerdo con el proceso Betterton-Kroll por tratamiento con calcio y magnesio metálicos, que forman una escoria de bismuto que pueden ser removida.46 Se puede obtener plomo muy puro procesando electrolíticamente el plomo fundido mediante el proceso de Betts. Dicho proceso utiliza ánodos de plomo impuro y cátodos de plomo puro en un electrolito constituido por una mezcla de fluorosilicato de plomo (PbSiF6) y ácido hexafluorosilícico (H2SiF6).46
El uso más amplio del plomo como tal se encuentra en la fabricación de acumuladores. Otras aplicaciones importantes son la fabricación de tetraetilo de plomo, forros para cables, elementos de construcción, pigmentos, soldadura suave, municiones, plomadas para pesca y también en la fabricación desde soldaditos de juguete hasta para hacer tubos de órganos musicales.
Se están desarrollando compuestos organoplúmbicos para aplicaciones como son la de catalizadores en la fabricación de espuma de poliuretano, tóxicos para las pinturas navales con el fin de inhibir la incrustación en los cascos, agentes biocidas contra las bacterias grampositivas, ácaros y otras bacterias, protección de la madera contra el ataque de los barrenillos y hongos marinos, preservadores para el algodón contra la descomposición y el moho, agentes molusquicidas, agentes antihelmínticos, agentes reductores del desgaste en los lubricantes e inhibidores de la corrosión para el acero.
Merced a su excelente resistencia a la corrosión, el plomo encuentra un amplio uso en la construcción, en particular en la industria química. Es resistente al ataque por parte de muchos ácidos porque forma su propio revestimiento protector de óxido, pero es atacado por las bases nitrogenadas. Como consecuencia de esta característica ventajosa, el plomo se utiliza mucho en la fabricación y el manejo del ácido sulfúrico, ácido nítrico.
Durante mucho tiempo se ha empleado el plomo como pantalla protectora para las máquinas de rayos X. En virtud de las aplicaciones cada vez más amplias de la energía atómica, se han vuelto cada vez más importantes las aplicaciones del plomo como blindaje contra la radiación.
Usos en el tiempo[editar]
En la historia[editar]

El plomo es uno de los metales más conocidos desde la antigüedad y el hombre lo empleó tanto por lo mucho que abunda como por su facilidad de fundirse. Suponen que Midácritas fue el primero que lo llevó a Grecia. Plinio el Viejo dice que en la antigüedad se escribía en láminas u hojas de plomo y algunos autores aseguran haber hallado muchos volúmenes de plomo en los cementerios romanos y en las catacumbas de los mártires. El uso de escribir en láminas de plomo es antiquísimo y Pausaniasmenciona unos libros de Hesíodo escritos sobre hojas de dicho metal. Se han encontrado en York (Inglaterra) láminas de plomo en que estaba grabada una inscripción del tiempo de Domiciano.8
En el Imperio romano las cañerías y las bañeras se recubrían con plomo o con cobre.
En la Edad Media se empleaban grandes planchas de plomo para las techumbres y para revestir la armazón de madera de las flechas o torres. También se fundían en plomo muchos medallones, mascarones de fuentes, etc. Y había también fuentes bautismales de plomo. En 1754 se halló en la Alcazaba o Albaicín de Granada una lámina de plomo de 30 pulgadas (76,2 cm) de largo y 4 (10,16 cm)de ancho con tres dobleces y entre ellos, una cruz y en 17 del mismo mes y año un libro de hojas de plomo escritas. Los caracteres de estos descubrimientos persuadieron de que eran de una fecha anterior al siglo VIII.8
En la actualidad[editar]
Su utilización como cubierta para cables, ya sea la de teléfono, de televisión, de internet o de electricidad, sigue siendo una forma de empleo adecuada. La ductilidad única del plomo lo hace muy apropiado para esta aplicación, porque puede estirarse para formar un forro continuo alrededor de los conductos internos.
El uso del plomo en pigmentos sintéticos o artificiales ha sido muy importante, pero está decreciendo en volumen. Los pigmentosque se utilizan con más frecuencia e intervienen en este elemento son:
- El blanco de plomo (conocido también como albayalde) 2PbCO3.Pb(OH)2
- Sulfato básico de plomo
- El tetróxido de plomo también conocido como minio.
- Cromatos de plomo.
- El silicatoeno de plomo (más conocido en la industria de los aceros blandos)
Se utilizan una gran variedad de compuestos de plomo, como los silicatos, los carbonatos y sales de ácidos orgánicos, como estabilizadores contra el calor y la luz para los plásticos de cloruro de polivinilo. Se usan silicatos de plomo para la fabricación de frituras (esmaltes) de vidrio y de cerámica, las que resultan útiles para introducir plomo en los acabados del vidrio y de la cerámica. La azida de plomo, Pb(N3)2, es el detonador estándar para los explosivos plásticos como el C-4 u otros tipos de explosivos H.E. (High Explosive). Los arseniatos de plomo se emplean en grandes cantidades como insecticidas para la protección de los cultivos y para ahuyentar insectos molestos como cucarachas, mosquitos y otros animales que posean un exoesqueleto. El litargirio (óxido de plomo) se emplea mucho para mejorar las propiedades magnéticas de los imanes de cerámica de ferrita de bario.
Asimismo, una mezcla calcinada de zirconato de plomo y de titanato de plomo, conocida como PETE, está ampliando su mercado como un material piezoeléctrico.
Efectos[editar]
Origen de la contaminación por plomo[editar]
Actualmente la mayor fuente de plomo es la atmósfera,9 aunque su contenido está disminuyendo gracias a la prohibición de utilizar gasolina con plomo. El plomo puede entrar en el agua potable a través de la corrosión de las tuberías. Esto es más común que ocurra cuando el agua es ligeramente ácida. Esta es la razón por la que los sistemas de tratamiento de aguas públicas ajustan el pH del agua potable. El plomo no cumple ninguna función esencial en el cuerpo humano y es muy dañino después de ser ingerido en la comida, o a través del aire o el agua.
Efectos en el organismo[editar]
El plomo puede causar varios efectos no deseados, como son:
- Perturbación de la biosíntesis de hemoglobina y subsecuentemente anemia
- Incremento de la presión sanguínea
- Daño a los riñones
- Aborto espontáneo
- Perturbación del sistema nervioso
- Daño al cerebro
- Disminución de la fertilidad del hombre a través del daño en el esperma
- Disminución de las habilidades de aprendizaje de los niños
- Perturbación en el comportamiento de los niños, como es agresión, comportamiento impulsivo e hipersensibilidad
- Alteraciones graves en la propiocepción, equilibriocepción, nocicepción y electrocepción, magnetocepción, ecolocalización en ciertos animales10
- La formación de depósitos plúmbicos en las encías que forman una línea de color gris claro azulado llamada "la línea del plomo" o "la línea de Burton"11
El plomo puede entrar en el feto a través de la placenta de la madre. Debido a esto puede causar serios daños al sistema nervioso y al cerebro de los niños por nacer.
Plomo en el medio ambiente[editar]
Con respecto a su incidencia en el medio ambiente, el plomo se encuentra de forma natural en el ambiente, pero las mayores concentraciones encontradas son el resultado de las actividades humanas.
Las sales de plomo entran en el medio ambiente a través de los tubos de escape (principalmente los defectuosos) de los coches, camiones, motos, aviones, barcos y aerodeslizadores y casi todos los tipos de vehículos motorizados que utilicen derivados del petróleo como combustible, siendo las partículas de mayor tamaño las que quedarán retenidas en el suelo y en las aguas superficiales, provocando su acumulación en organismos acuáticos y terrestres, y con la posibilidad de llegar hasta el hombre a través de la cadena alimenticia. Las pequeñas partículas quedan suspendidas en la atmósfera, pudiendo llegar al suelo y al agua a través de la lluvia ácida.
La acumulación de plomo en los animales puede causar graves efectos en su salud por envenenamiento, e incluso la muerte por paro cardio-respiratorio. Algunos organismos, como los crustáceos u otros invertebrados, son muy sensibles al plomo (dado que el plomo cuando se encuentra en exceso se deposita en los huesos y al no poseerlos queda retenido en su organismo), y en muy pequeñas concentraciones les causan graves mutaciones. Se registraron casos en donde las crías de crustáceos con saturnismo crónico, presentaban extremidades más largas, deformidades en otras y un comportamiento agresivo y poco coordinado llegando a producirse automutilaciones y autolaceraciones múltiples, atribuido a alteraciones genéticas generadas por la contaminación por plomo.
Otro efecto significativo del plomo en las aguas superficiales, es que provoca perturbaciones en el fitoplancton, que es una fuente importante de producción de oxígeno en los océanos y de alimento para algunos organismos acuáticos de variado tamaño (desde ballenas hasta pequeños pececillos).
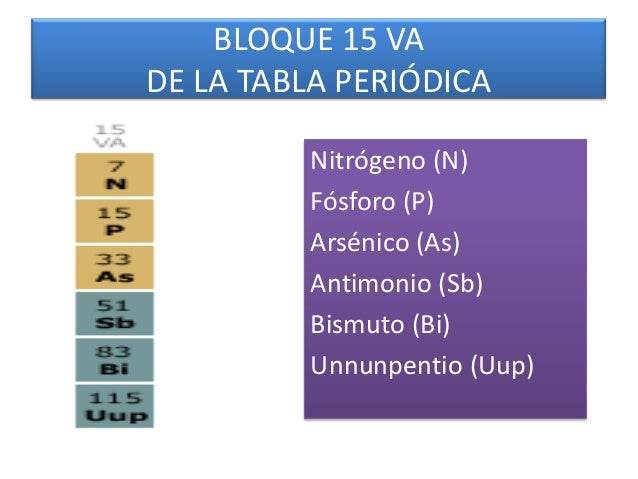
GRUPO 5A DE LA TABLA PERIÓDICA
El grupo del nitrógeno está compuesto por los elementos químicos del grupo 15 de la tabla periódica, que son: nitrógeno (N), fósforo (P), arsénico (As), antimonio(Sb), bismuto (Bi) y el elemento sintético moscovio (Mc), cuyo descubrimiento ya ha sido confirmado. Estos elementos también reciben el nombre de pnicógenos1 o nitrogenoideos.
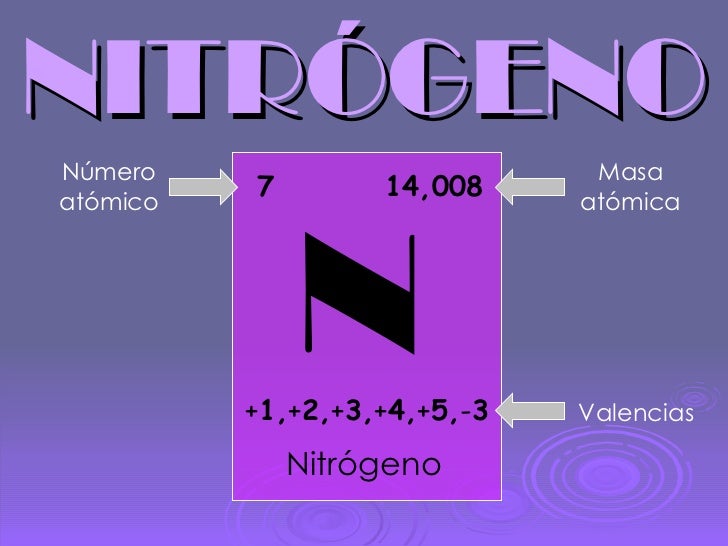
NITROGENO
El nitrógeno es un elemento químico de número atómico 7, símbolo N, su peso atómico es de 14,006 y que en condiciones normales forma un gas diatómico (nitrógeno diatómico o molecular) que constituye del orden del 78 % del aire atmosférico.1 En ocasiones es llamado ázoe (antiguamente se usó también Az como símbolo del nitrógeno).

Aplicación[editar]
La aplicación comercial más importante del nitrógeno diatómico es la obtención de amoníaco por el proceso de Haber. El amoníaco se emplea con posterioridad en la fabricación de fertilizantes y ácido nítrico.
Las sales del ácido nítrico incluyen importantes compuestos como el nitrato de potasio (nitro o salitre empleado en la fabricación de pólvora) y el nitrato de amonio fertilizante.
Los compuestos orgánicos de nitrógeno como la nitroglicerina y el trinitrotolueno son a menudo explosivos. La hidracinay sus derivados se usan como combustible en cohetes.
El ciclo de este elemento es bastante más complejo que el del carbono, dado que está presente en la atmósfera no solo como N2 (78 %) sino también en una gran diversidad de compuestos. Se puede encontrar principalmente como N2O, NO y NO2, los llamados NOx. También forma otras combinaciones con oxígeno tales como N2O3 y N2O5 (anhídridos), "precursores" de los ácidos nitroso y nítrico. Con hidrógeno forma amoníaco (NH3), compuesto gaseoso en condiciones normales.
Al ser un gas poco reactivo, el nitrógeno se emplea industrialmente para crear atmósferas protectoras y como gas criogénico para obtener temperaturas del orden de 78K de forma sencilla y económica. Inclusive se utiliza para inflar los neumáticos en los trenes de aterrizaje de los aviones, evitando condensación de agua a grandes alturas o su combustión al aterrizar.2
Etimología[editar]
Se considera que el nitrógeno (del latín nitrum -i, a su vez del griego νίτρον, "nitro" -nombre que históricamente se ha usado en forma vaga para referirse a diversos compuestos de sodio y de potasio que contienen nitrógeno-, y -geno, de la raíz griega γεν-, "generar"; es decir, "que genera salitre"3) fue descubierto formalmente por Daniel Rutherford en 1772, al dar a conocer algunas de sus propiedades (lo llamó "aire flogisticado", en función de lo que observó en su experimento de ese año4). Sin embargo, por la misma época también se dedicaron a su estudio Carl Wilhelm Scheele, quien lo aisló, Henry Cavendish y Joseph Priestley.
El nitrógeno es un gas tan inerte que Antoine Lavoisier se refería a él con el nombre azote (del griego ázoe, que significa "sin vida"56 (o tal vez lo llamó así por no ser apto para respirar7). Se clasificó entre los gases permanentes, sobre todo desde que Michael Faraday no consiguió verlo líquido a 50 atmósferas (atm) y –110 °C hasta los experimentos de Raoul Pictet y Louis Paul Cailletet, quienes en 1877 consiguieron licuarlo.
Los compuestos de nitrógeno ya se conocían en la Edad Media; así, los alquimistas llamaban aqua fortis al ácido nítricoy aqua regia (agua regia) a la mezcla de ácido nítrico y ácido clorhídrico, mezcla conocida por su capacidad para disolver el oro y el platino.
Abundancia y obtención[editar]
El nitrógeno es el componente principal de la atmósfera terrestre (78,1 % en volumen) y se obtiene para usos industriales de la destilación del aire líquido. Está presente también en los restos de animales, por ejemplo el guano, usualmente en la forma de urea, ácido úrico y compuestos de ambos. Por deficiencia causa falta de relajación de los músculos, problemas en el sistema cardiovascular, en el nervioso central y periférico.
También ocupa el 3 % de la composición elemental del cuerpo humano.
Se han observado compuestos que contienen nitrógeno en el espacio exterior y el isótopo Nitrógeno-14 se crea en los procesos de fusión nuclear de las estrellas. Se obtiene de Haizea
Compuestos[editar]
Con el hidrógeno forma el amoníaco (NH3), los nitritos (NO2), los nitratos (NO3), los ácidos nítricos (HNO3), la hidracina(N2H4) y el aziduro de hidrógeno (N3H, también conocido como azida de hidrógeno o ácido hidrazoico). El amoníaco líquido, anfótero como el agua, actúa como una base en una disolución acuosa, formando iones amonio (NH4+), y se comporta como un ácido en ausencia de agua, cediendo un protón a una base y dando lugar al anión amida (NH2). Se conocen largas cadenas y compuestos cíclicos de nitrógeno, pero son muy inestables.
Con los halógenos forma: NF3, NF2Cl, NFCl2, NCl3, NBr3.6 NH3, NI3.6 NH3, N2F4, N2F2 (cis y trans), N3F, N3Cl, N3Br y N3I.

Con el oxígeno forma varios óxidos que ya hemos nombrado: el nitroso o gas de la risa, el nítrico y el dióxido de nitrógeno. Son producto de procesos de combustión contribuyendo a la aparición de episodios contaminantes de smog fotoquímico. Otros óxidos son el trióxido de dinitrógeno (N2O3) y el pentóxido de dinitrógeno (N2O5), ambos muy inestables y explosivos.
Importancia biológica[editar]

El nitrógeno es un componente esencial de los aminoácidos y los ácidos nucleicos, vitales para los seres vivos. De todos los nutrientes minerales, el nitrógeno es el que mayor efecto tiene en el crecimiento de las plantas y, por lo tanto, en la productividad primaria de los ecosistemas, lo que afecta a su vez a todos los organismos que dependen de ellas:8 el aumento en el rendimiento de las cosechas a partir de que se comenzaron a utilizar fertilizantes nitrogenados en el siglo XIX lo demuestra.9 A pesar de la gran cantidad de nitrógeno atmosférico, este elemento es limitante: pocos organismos pueden asimilarlo en esta forma. Las plantas solamente pueden asimilarlo eficientemente forma de iones amonio (NH4+) o nitrato (NO3-), aunque también pueden absorber pequeñas cantidades de aminoácidos y urea.10
Algunas plantas han establecido relaciones simbióticas con hongos y procationtes capaces de reducir el nitrógeno atmosférico a amonio, a cambio de lo cual reciben moléculas energéticas de la planta hospedera. El nitrógeno reducido es así incorporado a la cadena trófica (véase también el ciclo del nitrógeno). Quizás el caso más conocido sea el de las bacterias del género Rhizobium con las leguminosas, pero también existen asociaciones con bacterias del género Frankia e inclusive algunas cianobacterias. Finalmente, también algunos hongos, llamados ectomicorrízicos, extienden sus filamentos más allá del alcance de las raíces, formando micorrizas que hacen más eficiente la absorción de nitritos, nitratos y amoniodel suelo en ambientes limitantes.11
Isótopos[editar]
Existen dos isótopos estables del nitrógeno, N-14 y N-15, siendo el primero —que se produce en el ciclo carbono-nitrógeno de las estrellas— el más común sin lugar a dudas (99,634 %). De los diez isótopos que se han sintetizado, uno tiene un periodo de semidesintegración de nueve minutos (el N-13), y el resto de segundos o menos.
Las reacciones biológicas de nitrificación y desnitrificación influyen de manera determinante en la dinámica del nitrógeno en el suelo, casi siempre produciendo un enriquecimiento en N-15 del sustrato.
Precauciones[editar]
Los fertilizantes nitrogenados son una importante fuente de contaminación del suelo y de las aguas. Los compuestos que contienen iones de cianuro forman sales extremadamente tóxicas y son mortales para numerosos animales, entre ellos los mamíferos.
Véase también: Nutrición de nitrógeno en plantas
Efectos del nitrógeno sobre la salud[editar]
Las moléculas de nitrógeno, en estado natural, se encuentran principalmente en el aire. En el agua y en los suelos el nitrógeno puede ser encontrado compuesto, en forma de nitratos y nitritos.
Los humanos han cambiado radicalmente las proporciones naturales de nitratos y nitritos, mayormente debido a la aplicación de estiércoles que contienen nitrato. El nitrógeno es emitido en grandes cantidades por las industrias. A lo largo de la historia, se nota un incremento de la presencia de nitratos y nitritos en el suelo y en el agua como consecuencia de reacciones que tienen lugar en el ciclo del nitrógeno. Esto se refleja en un incremento de la concentración de nitrógeno en las fuentes utilizadas para consumo humano, y por ende también en el agua potable.
Los nitratos y nitritos son conocidos por causar varios efectos sobre la salud humana. Estos son los efectos más comunes:12
- Tiene reacciones con la hemoglobina en la sangre, causando una disminución en la capacidad de transporte de oxígeno por la sangre. (nitrito)
- Provoca la disminución del funcionamiento de la glándula tiroidea. (nitrato)
- Ocasiona un bajo almacenamiento de la vitamina A. (nitrato)
- Favorece la producción de nitrosaminas, las cuales son conocidas como una de las causas más comunes de cáncer. (nitratos y nitritos)
Desde un punto de vista metabólico, el óxido de nitrógeno (NO) es mucho más importante que el nitrógeno. En 1987, Salvador Moncada descubrió que éste era un mensajero vital del cuerpo para la relajación de los músculos, y hoy se sabe que está involucrado en el sistema cardiovascular, el sistema inmunitario, el sistema nervioso central y el sistema nervioso periférico. La enzima que produce el óxido nítrico, la óxido-nítrico sintasa, es abundante en el cerebro.13
Aunque el óxido nítrico tiene una vida relativamente corta, se puede difundir a través de las membranas para llevar a cabo sus funciones. En 1991, un equipo encabezado por K.-E. Anderson del hospital universitario de Lund, Suecia, demostró que el óxido nítrico activa la erección por medio de la relajación del músculo que controla el flujo de sangre en el pene. La droga Viagra trabaja liberando óxido nítrico para producir el mismo efecto.
FOSFORO
El fósforo es un elemento químico de número atómico 15 y símbolo P. El nombre proviene del griego φώς [fos] ‘luz’ y φόρος [foros] ‘portador’. Es un no metal multivalente perteneciente al grupo del nitrógeno (Grupo 15 (VA): nitrogenoideos) que se encuentra en la naturaleza combinado en fosfatos inorgánicos y en organismos vivos pero nunca en estado nativo. Es muy reactivo y se oxida espontáneamente en contacto con el oxígeno atmosférico emitiendo luz.
El fósforo como molécula de Pi («fosfato inorgánico»), forma parte de las moléculas de ADN y ARN, las células lo utilizan para almacenar y transportar la energía mediante el adenosín trifosfato (ATP). Además, la adición y eliminación de grupos fosfato a las proteínas, fosforilación y desfosforilación, respectivamente, es el mecanismo principal para regular la actividad de proteínas intracelulares, y de ese modo el metabolismo de las células eucariotas tales como los espermatozoides.
Es un ciclo sedimentario, su reservorio es la corteza terrestre. El elemento se almacena en rocas fosfatadas y a medida que estas son erosionadas se van liberando compuestos fosfatados hacia el suelo y el agua. Luego son absorbidos por las plantas, a través de las raíces, incorporándose a los componentes vivos del sistema, a medida que pasan por los distintos niveles tróficos. Una vez que los organismos (plantas o animales) mueren, se descomponen y se libera el fósforo contenido en la materia orgánica.
Características principales[editar]

- El fósforo es un componente esencial de los organismos.
- Forma parte de los ácidos nucleicos (ADN y ARN).
- Forma parte de los huesos y dientes de los animales.
- En las plantas en una porción de 0,2 % y en los animales hasta el 1 % de su masa es fósforo.
- El fósforo común es un sólido.
- De color blanco, pero puro es incoloro.
- Un característico olor desagradable.
- Es un no metal.
- Emite luz por fosforescencia.
Existen varias formas alotrópicas del fósforo, siendo las más comunes el fósforo blanco y el rojo; ambos forman estructuras tetraédricas de cuatro átomos. El fósforo blanco, extremadamente tóxico e inflamable presenta dos formas, alfa y beta, con una temperatura de transición de −3,8 °C; expuesto a la luz solar o al calor (300 °C) se transforma en fósforo rojo en reacción exotérmica. Éste es más estable y menos volátil y tóxico que el blanco y es el que se encuentra normalmente en los laboratorios y con el que se fabrican las cerillas. El fósforo negro presenta una estructura similar al grafito y conduce la electricidad, es el más denso que los otros dos estados y no se inflama.
Función biológica[editar]
Los compuestos del fósforo intervienen en funciones vitales para los seres vivos, por lo que está considerado como un elemento químico esencial, aunque recientes experimentos apuntan que algunas formas de vida pudieran sustituirlo por arsénico. Forma parte de la molécula de Pi («fosfato inorgánico»), así como de las moléculas de ADN y ARN y de los fosfolípidos en las membranas lipídicas. Las células lo utilizan para almacenar y transportar la energía mediante el adenosín trifosfato. Además, la adición y eliminación de grupos fosfato a las proteínas, fosforilación y desfosforilación, respectivamente, es el mecanismo principal para regular la actividad de proteínas intracelulares, y de ese modo el metabolismo de las células eucariotas tales como los espermatozoides.
Historia[editar]
El fósforo —del latín phosphŏrus, y éste del griego φωσφόρος, portador de luz— antiguo nombre del planeta Venus, fue descubierto por el alquimista alemán Hennig Brand en 1669 en Hamburgo al destilar una mezcla de orina y arena (utilizó 50 cubos) mientras buscaba la piedra filosofal; al evaporar la urea obtuvo un material blanco que brillaba en la oscuridad y ardía con una llama brillante; desde entonces, las sustancias que brillan en la oscuridad sin emitir calor se las llama fosforescentes. Brand mantuvo su descubrimiento en secreto pero otro alquimista alemán, Kunckel, lo redescubrió en 1677 y enseñó a Boyle la forma de producirlo y aplicarlo.
Abundancia y obtención[editar]
Debido a su reactividad, el fósforo no se encuentra nativo en la naturaleza, pero forma parte de numerosos minerales. La apatita es una importante fuente de fósforo, existiendo importantes yacimientos en Marruecos, Rusia, Estados Unidos y otros países.
La forma alotrópica blanca se puede obtener por distintos procedimientos; en uno de ellos, el fosfato tricálcico, obtenido de las rocas, se calienta en un horno a 1450 °C en presencia de sílice y carbono reduciendo el fósforo que se libera en forma de vapor.

ARSÉNICO
El arsénico es un elemento químico de la tabla periódica que pertenece al grupo de los metaloides, también llamados semimetales, se puede encontrar de diversas formas, aunque raramente se encuentra en estado sólido.
Se conoce desde la antigüedad y se reconoce como extremadamente tóxico. A presión atmosférica el arsénico sublima a 613 °C.
Es un elemento esencial para la vida y su deficiencia puede dar lugar a diversas complicaciones, sin embargo, no se conoce con precisión, la función biológica.123 La ingesta diaria de 12 a 15 μg puede consumirse sin problemas en la dieta diaria de carnes, pescados, vegetales y cereales, siendo los peces y crustáceos los que más contenido de arsénico presentan.
El arsénico (del persa zarnikh, ‘oropimente amarillo’ o bien del griego arsenikón, ‘masculino’) es un elemento químico de la tabla periódica cuyo símbolo es As y el número atómico es 33. En la tabla periódica de los elementos se encuentra en el quinto grupo principal. El arsénico se presenta raramente sólido, principalmente en forma de sulfuros. Pertenece a los metaloides, ya que muestra propiedades intermedias entre los metales de transición y los no metales.
Se conocen compuestos de arsénico desde la antigüedad, siendo extremadamente tóxicos, aunque se emplean como componentes en algunos medicamentos. El arsénico es usado para la fabricación de semiconductores y como componente de semiconductores III-V como el arseniuro de galio.
El arsénico es muy común en la atmósfera terrestre, en rocas y suelos, en la hidrosfera y la biosfera. Es llevado al ambiente a través de una combinación de procesos como:
- Naturales como la meteorización, actividad biológica, emisiones volcánicas
- Antropogénicos como la actividad minera, uso de combustibles fósiles, uso de pesticidas, herbicidas, etc.
Características principales[editar]

El arsénico se presenta en tres estados alotrópicos, gris o metálico, amarillo y negro.4 El arsénico gris metálico (forma α) es la forma estable en condiciones normales y tiene estructura romboédrica, es un buen conductor del calor pero pobre conductor eléctrico, su densidad es de 5,73 g/cm³, es deleznable y pierde el lustre metálico expuesto al aire.5
El arsénico “amarillo” (forma γ) se obtiene cuando el vapor de arsénico se enfría muy rápidamente. Es extremadamente volátil y más reactivo que el arsénico metálico y presenta fosforescencia a temperatura ambiente. El gas está constituido por moléculas tetraédricas de As4 de forma análoga al fósforoy el sólido formado por la condensación del gas tiene estructura cúbica, es de textura jabonosa y tiene una densidad aproximada de 1,97 g/cm³.6 Expuesto a la luz o al calor revierte a la forma estable (gris). También se denomina arsénico amarillo al oropimente, mineral de trisulfuro de arsénico.
Una tercera forma alotrópica, el arsénico “negro” (forma β) de estructura hexagonal y densidad 4,7 g/cm³, tiene propiedades intermedias entre las formas alotrópicas descritas y se obtiene en la descomposición térmica de la arsina o bien enfriando lentamente el vapor de arsénico.
Todas las formas alotrópicas excepto la gris carecen de lustre metálico y tienen muy baja conductividad eléctrica por lo que el elemento se comportará como metal o no metal en función, básicamente, de su estado de agregación.7 También vea metal pesado.
A presión atmosférica el arsénico sublima a 613 °C, y a 400 °C arde con llama blanca formando el sesquióxido As4O6. Reacciona violentamente con el cloro y se combina, al calentarse, con la mayoría de los metales para formar el arseniuro correspondiente y con el azufre. No reacciona con el ácido clorhídrico en ausencia de oxígeno, pero sí con el nítrico caliente, sea diluido o concentrado y otros oxidantes como el peróxido de hidrógeno, ácido perclórico, etc. Es insoluble en agua pero muchos de sus compuestos lo son.
Es un elemento químico esencial para la vida aunque tanto el arsénico como sus compuestos son extremadamente venenosos.6
Se encuentra en el 2.º grupo analítico de cationes; precipita con H2S de color amarillo.
Aplicaciones[editar]
En uso[editar]
- Preservante de la madera (arseniato de plomo y cromo), uso que representa, según algunas estimaciones, cerca del 70 % del consumo mundial de arsénico.
- El arseniuro de galio es un importante material semiconductor empleado en circuitos integrados más rápidos, y caros, que los de silicio. También se usa en la construcción de diodos láser y LED.
- Aditivo en aleaciones de plomo y latones.
- Insecticida (arseniato de plomo), herbicidas (arsenito de sodio) y venenos: a principios del siglo XX se usaban compuestos inorgánicos pero su uso ha desaparecido prácticamente en beneficio de compuestos orgánicos (derivados metílicos). Sin embargo, esas aplicaciones están declinando.8
- El disulfuro de arsénico se usa como pigmento y en pirotecnia.
- Decolorante en la fabricación del vidrio (trióxido de arsénico).
En desuso[editar]
- Históricamente el arsénico se ha empleado con fines terapéuticos prácticamente abandonados por la medicina occidental6 aunque recientemente se ha renovado el interés por su uso como demuestra el caso del trióxido de arsénico para el tratamiento de pacientes con leucemia promielocítica aguda.9
- Como elemento fertilizante en forma de mineral primario rico, para la agricultura.
- A lo largo de la historia el arsénico y sus compuestos han sido utilizados con fines homicidas, fundamentalmente en forma de anhídrido arsenioso (polvo blanco, insípido e inodoro llamado rey de los venenos).
- Elaboración de insecticidas, herbicidas, raticidas, fungicidas, etc, aunque cada vez se utiliza menos con estos fines.
Función biológica[editar]

Si bien el arsénico se asocia con la muerte, es un elemento esencial para la vida y su deficiencia puede dar lugar a diversas complicaciones.4 La ingesta diaria de 12 a 15 μg puede obtenerse sin problemas con la dieta diaria de carnes, pescados, vegetales y cereales, siendo los peces y crustáceos los que más contenido de arsénico presentan, generalmente en forma de arsenobetaína, menos tóxica que el arsénico inorgánico.
El 2 de diciembre de 2010, la Agencia Espacial Estadounidense (NASA) confirmó el hallazgo de la Dra. Felisa Wolfe-Simonsen del Instituto de Astrobiología de la NASA, en las aguas tóxicas y salobres del Lago Mono, en California, una bacteria de la familia Halomonadaceae que puede sustituir el fósforo (que hasta la fecha se consideraba indispensable para la vida) con arsénico, al punto de incorporar este elemento a su ácido desoxirribonucleico (ADN).10 Este descubrimiento abre la puerta a la búsqueda de nuevas formas de vida en planetas que no contengan fósforo en su atmósfera. Sin embargo, en un estudio realizado en 2012 algunos de los descubrimientos fueron refutados.11 Aparentemente, la bacteria sí es resistente al arsénico pero no puede sustituir por completo el fósforo.
Si bien la información anterior fue publicada en la prestigiosa revista científica Science, a la fecha los resultados han y siguen siendo fuertemente cuestionados por numerosos científicos que han tratado de reproducir el mismo diseño experimental sin resultados positivos, a raíz de lo cual han postulado que la bacteria GFAJ-1 pudo sobrevivir en el medio de cultivo sintético empleado para la experimentación gracias a las trazas de fósforo presentes en él.12
Historia[editar]

El arsénico (del griego άρσενιχόν, oropimente) se conoce desde tiempos remotos, lo mismo que algunos de sus compuestos, especialmente los sulfuros. Dioscórides y Plinio el Viejo (griegos siglo I) conocían las propiedades del oropimente y el rejalgar y Celso Aureliano (romano siglo I), Galeno(siglo II) sabían de sus efectos irritantes, tóxicos, corrosivos y parasiticidas y observaron sus virtudes contra las toses pertinaces, afecciones de la voz y las disneas.
Los médicos árabes usaron también los compuestos de arsénico en fumigaciones, píldoras y pociones además de en aplicaciones externas. Durante la Edad Media los compuestos arsenicales cayeron en el olvido quedando relegados a los curanderos que los prescribían contra la escrófula y el hidrocele.
Roger Bacon y Alberto Magno se detuvieron en su estudio —se cree que este último fue el primero en aislar el elemento en el año 1250— y Paracelso hizo de él una panacea. Leonardo da Vinci lo utilizó mediante endoterapia aplicándolo a los manzanos para controlar a los ladrones de frutas.
El primero que lo estudió con detalle fue Brandt en 1633 y Schroeder lo obtuvo en 1649 por la acción del carbón sobre el ácido arsénico. A Berzeliuss se deben las primeras investigaciones acerca de la composición de los compuestos del arsénico.
En el siglo XVIII los arsenicales consiguieron un puesto de primer orden en la terapéutica hasta que fueron sustituidos por las sulfamidas y los antibióticos.
Abundancia y obtención[editar]

Es el 52.º elemento en abundancia de la corteza terrestre con 2 ppm (5·10−4 %) y es uno de los 22 elementos conocidos que se componen de un solo núcleo estable. El arsénico se encuentra en forma nativa y, principalmente, en forma de sulfuro en una gran variedad de minerales que contienen cobre, plomo, hierro (arsenopirita o mispickel), níquel, cobalto y otros metales.
En la fusión de minerales de cobre, plomo, cobalto y oro se obtiene trióxido de arsénico que se volatiliza en el proceso y es arrastrado por los gases de la chimenea que pueden llegar a contener más de una 30 % de trióxido de arsénico. Los gases de la chimenea se refinan posteriormente mezclándolos con pequeñas cantidades de galena o pirita para evitar la formación de arsenitos y por tostación se obtiene trióxido de arsénico entre el 90 y 95 % de pureza, por sublimaciones sucesivas puede obtenerse con una pureza del 99 %.
Reduciendo el óxido con carbón se obtiene el metaloide, sin embargo la mayoría del arsénico se comercializa como óxido. Prácticamente la totalidad de la producción mundial de arsénico metálico es de China, que es también el mayor productor mundial de trióxido de arsénico.
Según datos del servicio de prospecciones geológicas estadounidense (U.S. Geological Survey) las minas de cobre y plomo contienen aproximadamente 11 millones de toneladas de arsénico, especialmente en Perú y Filipinas, y el metaloide se encuentra asociado con depósitos de cobre-oro en Chile y de oro en Canadá.
También es un componente del tabaco y es altamente tóxico.
Precauciones[editar]
El arsénico y sus compuestos son extremadamente tóxicos, especialmente el arsénico inorgánico. En Bangladés se ha producido una intoxicación masiva, la mayor de la historia, debido a la construcción de infinidad de pozos de agua promovida por las ONG occidentales que han resultado estar contaminados afectando a una población de cientos de miles de personas.13 También otras regiones geográficas, España incluida, se han visto afectadas por esta problemática.

ANTIMONIO
El antimonio es un elemento químico que forma parte del grupo de los metaloides de número atómico 51 situado en el grupo 15 de la tabla periódica de los elementos. Su nombre y abreviatura (Sb) procede de estibio, término hoy ya en desuso, que a su vez procede del latín stibium ("Banco de arena gris brillante"), de donde se deriva la palabra estibio.note 1 Su principal mena es la estibina.
Este elemento semimetálico tiene cuatro formas alotrópicas. En su forma estable es un metal blanco azulado. El antimonio negro y el amarillo son formas no metálicas inestables. Principalmente se emplea en aleaciones metálicas y algunos de sus compuestos para dar resistencia contra el fuego, en pinturas, cerámicas, esmaltes, vulcanización del caucho y fuegos artificiales.
Características principales[editar]
El antimonio en su forma elemental es un sólido cristalino, fundible, quebradizo, blanco plateado que presenta una conductividad eléctrica y térmica baja y se evapora a bajas temperaturas. Este elemento semimetálico se parece a los metales en su aspecto y propiedades físicas, pero se comportan químicamente como un no metal. También puede ser atacado por ácidos oxidantes y halógenos.
Las estimaciones sobre la abundancia de antimonio en la corteza terrestre van desde 0,2 a 0,5 ppm. El antimonio es calcófilo, presentándose con azufre y con otros elementos como plomo, cobre y plata.1
Historia[editar]
_Diego_Mateo_Zapata.jpg)
Estudios arqueológicos e históricos indican que el antimonio y sus sulfuros han sido usados por los humanos al menos durante los últimos 6 milenios. En la antigüedad la antimonita o estibina, Sb2S3, la forma más común de sulfuro de antimonio fue el principal ingrediente del “kohl”, una pasta negra usada por los egipcios, entre otros, como maquillaje para los ojos.23 Los babilonios conocían la forma de obtener antimonio de sus compuestos y lo usaban como ornamento para vasijas.
El alquimista Basil Valentine (1565-1624), presentado a veces como el descubridor del antimonio, fue el primero en describir la extracción de antimonio de sus compuestos en su tratado “Triumph Wagens des Antimonij” (El carro triunfal del antimonio).4
El nombre antimonio viene de una latinización de la palabra árabe انتيمون ("al-ithmīd"), que a su vez consistía en una arabización de la palabra latinastibium.567
Otras teorías sugieren que antimonio es un compuesto de las palabras latinas “anti"(miedo) y “mono” (solo); lo que haría referencia a su existencia en la naturaleza normalmente como compuesto.note 289
Tras la invención de la imprenta en el siglo XVI el antimonio fue usado como aleante para los sellos tipográficos. Al enfriar, el antimonio líquido tiene la propiedad excepcional de expandirse mientras se solidifica. De este modo consigue rellenar las grietas de los moldes, por lo que las aristas de las piezas que se obtienen son muy afiladas. Por esta razón, se usó para hacer tipos de imprenta. En el siglo XIX su aleación con zinc (metal inglés) fue utilizada en los cubiertos, palmatorias y candelabros.
Tras el invento del acumulador eléctrico se comprobó que el uso de la aleación de plomo y antimonio hacía durar mucho más a los mismos. Durante la Primera Guerra Mundial se alcanzó un máximo de producción, debido a su uso armamentístico, ya que este semimetal aumenta mucho la dureza y la fuerza mecánica del plomo y del estaño. Con el desarrollo de la industria automovilística el uso del antimonio ha ido aumentando año tras año, aunque los niveles de la Primera Guerra Mundial no se volvieron a alcanzar hasta los años 1990.
Aplicaciones[editar]
El antimonio tiene una creciente importancia en la industria de semiconductores en la producción de diodos, detectores infrarrojos y dispositivos de efecto Hall.10
Usado en aleaciones, este semimetal incrementa mucho la dureza y resistencia a esfuerzos mecánicos de la aleación. También se emplea en distintas aleaciones como peltre, metal antifricción (aleado con estaño), metal inglés (formado por zinc y antimonio), etc.11
Algunas aplicaciones más específicas:
- Baterías y acumuladores12
- Tipos de imprenta131415
- Recubrimiento de cables
- Cojinetes y rodamientos
Compuestos de antimonio en forma de óxidos, sulfuros, antimoniatos y halogenuros de antimonio se emplean en la fabricación de materiales resistentes al fuego, esmaltes, vidrios, pinturas y cerámicas.16 El trióxido de antimonio es el más importante y se usa principalmente como retardante de llama.17 Estas aplicaciones como retardantes de llama comprenden distintos mercados como ropa, juguetes, o cubiertas de asientos.1819
Usos metálicos[editar]

Desde que la batería eléctrica de plomo y ácido fue desarrollada en el siglo XIX, ha sido en gran medida la batería secundaria (o recargable) más importante por todo el mundo. Se utilizan en vehículos de motor, o como baterías industriales.
Las baterías eléctricas industriales incluyen las baterías de acumuladores de tracción en las locomotoras de las minas, carros del golf, y así sucesivamente, baterías de "energía de emergencia". El antimonio en aleación con el plomo es usado para ciertas piezas de los acumuladores eléctricos para las cuales la resistencia a la corrosión es necesaria.
El antimonio es un componente menor pero importante de muchas soldaduras suaves, que son las soldaduras que funden en temperaturas debajo del 625 K. Estas soldaduras pueden contener entre 0,5 y 3% de antimonio. La función del antimonio en estas soldaduras es consolidar la soldadura y suprimir la formación del alótropos de estaño a bajas temperatura, lo que degradaría de otra manera la integridad estructural de los empalmes soldados en las temperaturas debajo del punto de la transición de fase (289 K). El antimonio se ha utilizado como un endurecedor para el plomo usado en la munición.
En los Estados Unidos su uso se confina en gran parte a la fabricación de balas y perdigones. La contaminación del agua subterránea, del suelo y de la cadena tróficacon el tóxico plomo ha preocupado por muchos años, y las regulaciones ambientales han conducido al reemplazo del plomo al antimonio con una aleación de tungsteno.
Las aleaciones de plomo que contienen cerca del 2 al 8% de antimonio son resistentes al uso atmosférico y la corrosión por lo que son utilizadas en la construcción de canales y barreras de la humedad. En la industria química, las aleaciones que contienen a partir 4 a 15 % de antimonio proporcionan la protección contra varios estados líquidos de los productos químicos, especialmente del ácido sulfúrico o del azufre. Aleado con bismuto, plomo y estaño, el antimonio es un componente de algunas de las aleaciones fusibles usadas en dispositivos de seguridad de fuego. El metal que se emplea para la fabricación de caracteres y demás material tipográfico se obtiene con una aleación de plomo, antimonio y estaño. El plomo se usa por la fácil fusión y para que la aleación sea dúctil y compacta. El antimonio sirve para dar más resistencia al metal con el fin de que no se aplaste tan fácilmente durante las repetidas y numerosas tiradas. Las aleaciones son diversas, según los tamaños de los tipos y el uso a que se destinan.
Así que para la fabricación del metal destinado a blancos, se suele usar la aleación siguiente, denominada ordinaria: 75 partes de plomo, 20 partes de antimonio y 5 partes de estaño. Cantidades pequeñas de antimonio de gran pureza se utilizan en los vídeo discos (DVD).
Usos no metálicos[editar]
La punta de los fósforos de seguridad contiene trisulfuro de antimonio. La combustión es una reacción exotérmica mantenida por los radicales libres internamente generados y el calor radiante. Los retardadores con halógeno de la llama actúan interfiriendo con el mecanismo de cadena radical en la fase de gas (la llama). Cuando son utilizados por sí mismos, los retardadores de la llama del halógeno se deben utilizar en cantidades muy grandes. Este problema se evita agregando el trióxido del antimonio, que trabaja de forma conjunta con los halógenos, reduciendo la cantidad necesaria de retardante de llama y reduciendo también el coste del tratamiento total. El mecanismo del trabajo conjunto del antimonio y los halógenos se ha intentado explicar de varias maneras, pero ninguna es definitiva.
Muchos plásticos comunes son susceptibles a la degradación por el calor y la luz ultravioleta (UV) y se deben proteger durante la vida de servicio los productos hechos de ellos por la adición de compuestos conocidos como estabilizadores. El antimonio ha sido utilizado desde los años 1950 como estabilizador de calor eficaces para el PVC, especialmente en las formas rígidas del plástico.
El trióxido de antimonio se utiliza como catalizador en la polimerización del PET, que es un plástico usado en las botellas, películas, acondicionamiento de los alimentos, y muchos otros productos. Los compuestos del antimonio, junto con el dióxido de germanio, son los catalizadores preferidos para PET.
El dióxido de germanio da un producto con una transparencia mejor que el antimonio, pero que es demasiado costoso para muchas aplicaciones del PET. El trióxido de antimonio es utilizado también como pigmento blanco para las pinturas exteriores, donde su resistencia al desgaste por la acción atmosférica le hizo el objeto de valor, sin embargo, al descubrirse su capacidad tóxica el trióxido de antimonio ha sido suplantado por el dióxido de titanio (TiO2).
Todavía se utiliza en cantidades significativas como estabilizador del color, donde es importante mantener intensidad del color y evitar el cambio de la tonalidad, por ejemplo en las pinturas amarillas usadas para los autobuses de las escuelas (estadounidenses y sudafricanas) y en las rayas amarillas aplicadas a los pavimentos del camino.
Los pigmentos conductores de la electricidad del óxido de estaño (SnO) con antimonio se han introducido en años recientes para incorporarlos en las capas plásticas que protegen las computadoras y otros componentes electrónicos contra la electricidad estática.
El antimonio fue utilizado en medicina, por su buenas cualidades expectorantes, eméticas y purgantes. Y se llegaron a escribir tratados sobre sus cualidades médicas. Hasta que se decidió declararlo veneno, de forma oficial, el 3 de agosto de 1866. En forma de sales de antimonio Sb(OH)2Cl (Sbv) o como antimonio pentavalente, aún se menciona como tratamiento inicial contra leishmaniasis.
Antimonio y ambiente[editar]
El antimonio es liberado al ambiente desde fuentes naturales e industriales. Puede permanecer en el aire adherido a partículas muy pequeñas por muchos días. La mayoría del antimonio en el aire se deposita en el suelo, en donde se adhiere firmemente a partículas que contienen hierro, manganeso o aluminio. Altos niveles de antimonio en el aire que respiramos por períodos muy largos pueden ocasionar irritación de los ojos y los pulmones y causar problemas respiratorios, del corazón y del estómago.
El límite de exposición ocupacional es 0,5 mg de antimonio por m3 de aire por un día laborable de 8 h. El nivel máximo permitido del antimonio en agua potable en Europa es 0,006 ppm.
En el aire urbano, la principal fuente de antimonio es la combustión de combustibles fósiles en vehículos automotores, centrales eléctricas e incineradores.
El inventario tóxico de Estados Unidos de la Agencia de Protección del Ambiente (EPA) para el período a partir de 1993 a 2005 demostró que las plantas industriales de E. U. A. lanzaron más de 900 t/año de antimonio en todas las formas a la tierra y cerca de 25 t/año al agua subterránea. Del antimonio lanzado a la tierra por industrias importantes, los fundidores de cobre primarios suponen cerca de 60 %; fundidores primarios para otros metales no ferrosos, 20%; fundidores no ferrosos secundarios, 7% y refinerías de petróleo, 2%. El 11% restante se atribuye a la fabricación de varios productos del antimonio. El lanzamiento postconsumición del antimonio de productos desechados del uso final es también de importancia.
Hay preocupación, especialmente en Europa, por la lixiviación de los pigmentos del antimonio, de los estabilizadores de calor, y de los retardadores de la llama de productos desechados de los plásticos. Estas preocupaciones han contribuido a un cambio a los estabilizadores de calcio-cinc en Europa y a los estabilizadores basados en estaño en Estados Unidos y el Japón. Se cree que el país que más antimonio lanza a la atmósfera es China, debido a gran uso que se hace de este elemento en ese país, ya que posee las principales minas de antimonio del mundo. Sin embargo, debido al régimen político no se tienen datos.
Abundancia y obtención[editar]
El antimonio se encuentra en la naturaleza en numerosos minerales, aunque es un elemento poco abundante. Pero es posible encontrarlo libre, normalmente está en forma de sulfuros; la principal mena de antimonio es la antimonita (también llamada estibina), Sb2S3.20
Mediante el tostado del sulfuro de antimonio se obtiene óxido de antimonio (III), Sb2O3, que se puede reducir con coque para la obtención de antimonio.
- 2Sb2O3 + 3C → 4Sb + 3CO2
También se puede obtener por reducción directa del sulfuro, por ejemplo con chatarra de hierro:
- Sb2S3 + 3Fe → 2Sb + 3FeS
Compuestos[editar]
Sus estados de oxidación más comunes son el 3 y el 5.
Los términos antimonio crudo y crudum se aplican al mineral que contiene más de 90 por ciento de antimonio, y al mineral del sulfuro licuado, que es esencialmente una mezcla del antimonio-sulfuro que contiene 70 por ciento o más antimonio. El metal refinado del antimonio es la forma común estable de antimonio.[cita requerida]
El antimonio amarillo o alfa-antimonio se produce por la acción de ozono en SbH3 líquido, -90 °C. Es amorfo y poco soluble en disulfuro de carbono. El antimonio amarillo es muy inestable y se transforma fácilmente a temperaturas superiores -90 °C en antimonio negro, que también puede formarse directamente a partir de SbH3líquido y oxígeno a -40 °C. El antimonio negro se oxida espontáneamente en aire y se convierte en el antimonio romboédrico ordinario o beta-antimonio. La cuarta forma alotrópica del antimonio es el antimonio explosivo, que se forma a partir de la electrólisis del cloruro de antimonio.[cita requerida]
Esta forma se transforma a 475 K en la forma alotrópica más común produciendo una explosión. Hay estudios que intentan demostrar que el antimonio amarillo es en realidad antimonio impuro y no es una forma alotrópica verdadera del antimonio.
Debido a su dureza, fragilidad, y carencia del maleabilidad, el antimonio no tiene ninguna aplicación como metal por sí mismo a excepción de las cantidades pequeñas usadas para los bastidores ornamentales y los dispositivos de semiconductor. Sin embargo, es un componente de menor importancia en muchas aleaciones del plomo y estaño.
La mayoría del antimonio que se utiliza en el estado metálico, como en baterías del LA, la cubierta del cable, y varios otros usos, se utiliza como cierta forma de plomo antimonial, que puede contener hasta 25% de antimonio, pero contiene más comúnmente porcentajes de un solo dígito. El antimonio es también un componente de varias aleaciones de estaño, tales como metal de bretaña, metal antifricción y soldaduras de estaño-antimonio-plata usada para ensamblar tubos para agua potable.
El antimonio forma un número muy grande de compuestos inorgánicos. Los sulfuros predominan en naturaleza y están disponibles para el comercio como minerales procesados del antimonio. En términos de las cantidades producidas, el compuesto sintético más importante del antimonio en gran medida es el trióxido (Sb2O3), que es utilizado por sí mismo para algunas aplicaciones.
Otros compuestos usados en cantidades substanciales son el pentóxido (Sb2O5), el trisulfuro (Sb2S3) y el pentasulfuro (Sb2S5). Estos compuestos se utilizan como los retardadores de la llama, en los pigmentos, estabilizadores del calor y de la radiación en los plásticos y de catalizadores.
Se conocen todos sus trihalogenuros, SbX3, y el pentafluoruro y pentacloruro, SbX5. El trifluoruro se emplea como fluorante. El pentafluoruro junto con HSO3F forma un sistema SbF5-FSO3H con propiedades de superácido. Con estos halogenuros se pueden preparar distintos complejos. Se conoce el hidruro SbH3 (estibina), pero es poco estable y se descompone con mucha facilidad.
Se conoce el trióxido de antimonio, Sb2O3 y el pentóxido, Sb2O5.
Precauciones[editar]
El antimonio y muchos de sus compuestos son tóxicos, debiéndose tener los mayores cuidados posibles en su manipulación. Reacciona violentamente con oxidantes fuertes (ejemplo: halógenos, permanganatos alcalinos y nitratos) originando riesgo de incendio y explosión. Reacciona en medio ácido con hidrógeno naciente produciendo un gas muy tóxico (estibamina). En contacto con ácidos concentrados en caliente, emite gases tóxicos (estibamina). Estos compuestos se forman en presencias de metales atacables por el ácido que se está usando, como por ejemplo el hierro, por lo que nunca deben emplearse objetos metálicos (recipientes, pinzas, etc.) cuando se limpien con ácido minerales de antimonio.2122
Su temperatura de autoignición es 900 °C, y su almacenamiento debe realizarse separado de alimentos y piensos, oxidantes fuertes, ácidos, sustancias reductoras.2324 Se debe manejar con guantes, gafas protectoras.

BISMUTO
El bismuto es un elemento químico de la tabla periódica cuyo símbolo es Bi, su número atómico es 83 y se encuentra en el grupo 15 del sistema periódico.
Ya era conocido en la antigüedad, pero hasta mediados del siglo XVIII era confundido con el plomo, estaño y zinc. Ocupa el lugar 73 en abundancia entre los elementos de la corteza terrestre (representa el 8,5x10-7 % del peso de la corteza) y es tan escaso como la plata. Los principales depósitos están en Sudamérica, pero en Estados Unidos se obtiene principalmente como subproducto del refinado de los minerales de cobre y plomo.
Es un metal típico desde el punto de vista químico. En compuestos, tiene valencias de +3 (bismuto (III)) o +5 (bismuto (V)), siendo más estables los compuestos de bismuto trivalente. Existen varios nitratos, especialmente el nitrato de bismuto, Bi(NO3)3, o trinitrato de bismuto, y su pentahidrato, Bi(NO3)3•5H 2O, que se descompone en nitrato de bismuto. Éste también se conoce como oxinitrato de bismuto, nitrato de bismutilo, blanco perla y blanco de España, y se emplea en medicina y en cosmética.
El bismuto se expande al solidificarse; esta extraña propiedad lo convierte en un metal idóneo para fundiciones. Algunas de sus aleaciones tienen puntos de fusión inusualmente bajos. Es una de las sustancias más fuertemente diamagnéticas (dificultad para magnetizarse). Es un mal conductor del calor y la electricidad, y puede incrementarse su resistencia eléctrica en un campo magnético, propiedad que lo hace útil en instrumentos para medir la fuerza de estos campos. Es opaco a los rayos X y puede emplearse en fluoroscopia.
Entre los elementos no radiactivos, el bismuto tiene el número atómico y la masa atómica (208,98) más altos. Tiene un punto de fusión de 271 °C, un punto de ebullición de 1560 °C y una densidad de 9800 kg/m³.

Historia

El bismuto es uno de los primeros diez metales que fueron descubiertos, ya conocido desde la antigüedad, por lo que a ninguna persona se le atribuye su descubrimiento. El elemento se confundió en los primeros tiempos con el estaño y el plomo, debido a su parecido con esos elementos.Georgius Agricola, en De Natura Fossilium (ca. 1546) afirma que el bismuto es un metal distinto en una familia de metales que incluía al estaño y al plomo, basándose en la observación de sus propiedades físicas.1 Los mineros en la edad de la alquimia también dieron al bismuto el nombre de tectum argenti, o "plata haciéndose", en el sentido de que la plata estaría todavía en proceso de formación dentro de la Tierra.234
A partir de Johann Heinrich Pott en 1738,5 Carl Wilhelm Scheele y Torbern Olof Bergman, la distinción entre el plomo y el bismuto se hizo evidente, y Claude François Geoffroy demostró en 1753 que este metal era distinto del plomo y del estaño.367
El bismuto también era conocido por los incas y fue utilizado (junto con el habitual cobre y estaño) en una aleación de bronce especial para cuchillos.8
El nombre bismuto es de etimología incierta. Aparece en la década de 1660, a partir de los términos obsoletos alemanes, Bismuth, Wismut o Wissmuth (inicios del siglo XVI); tal vez relacionado con el antiguo alto alemán hwiz("blanco").9 El nuevo latín bisemutum (debido a Agricola, que latinizó muchas palabras mineras y técnicas alemanas) es del alemán Wismuth, tal vez del weiße Masse, "masa blanca".10
Características del bismuto[editar]

Cuando es sólido flota sobre su estado líquido, por tener menor densidad en el estado sólido. Esta característica es compartida con el agua, el galio, el ácido acético, el antimonio y el silicio.
En casi todos los compuestos de bismuto aparece en forma trivalente, no obstante, en ocasiones puede ser pentavalente o monovalente. El bismutato de sodio y el pentafluoruro de bismuto son quizá los compuestos más importantes de Bi(V). El primero es un agente oxidante poderoso y el último un agente fluorante útil para compuestos orgánicos.
El átomo de bismuto se sigue considerando popularmente como el más pesado entre los átomos estables, ya que su tiempo de vida es varios millones la edad total del Universo, además de que, en teoría, todos los elementos químicos a partir del niobio están sujetos a fisión espontánea, es decir, todos los elementos con número superior al número 41 teóricamente pueden ser inestables, si bien en el bismuto la desintegración fue observado por estudios franceses en la última década. Es también el elemento no radiactivo monoatómico más pesado que existe.
El bismuto es uno de los dos peores conductores térmicos que existen entre todos los metales (junto al manganeso); es también el metal más diamagnético y sus aleaciones aprovechan ambas ventajas en situaciones donde se requiera. No existe de manera natural en el cuerpo humano ni en ninguna forma de vida en general. Se utiliza mucho en medicina, siendo parte de los astringentes recetados para problemas relacionados con el sistema digestivo, diarreas fuertes o irritaciones esofágicas, del colon, duodeno o intestinos.
Químicamente recuerda a los metales nobles y preciosos, se oxida con dificultad y se mantiene en algunos ácidos como el clorhídrico. Puede presentarse en estado nativo, hecho que refuerza su nobleza. El metal es gris con un muy ligero toque rosado, muy «vidrioso» y frágil, no soporta un impacto mínimo, su ductilidad y maleabilidad es nula. De no ser por su escasez, podría reemplazar al plomo como escudo antinuclear debido a la gran masa atómica que posee.
El bismuto se considera un metal pesado pero es irónicamente muy poco tóxico, prácticamente no agresivo, pese a estar rodeado de metales venenosos y peligrosos para el medioambiente. Sus cristales pueden ser trabajados hasta conseguir resultados de una increíble belleza. Oxidado en el laboratorio se consiguen maclas de iris fascinantes.
«El metal es muy caro teniendo en cuenta su escasez (igual a la del oro) y dificultad para encontrarlo. No parece demasiado importante en ningún sector de la industria o la medicina, pues se usa muy poco.»
El bismuto será el último elemento en desintegrarse en el universo. La vida media del elemento se estima en 20 trillones de años.11
Aplicaciones[editar]
Sustituto del plomo[editar]
La diferencia entre las densidades del plomo (densidad 11.32 g·cm−3) y del bismuto (densidad 9.78 g·cm−3) es lo suficientemente pequeña para que pueda ser utilizado en lugar del plomo en numerosos usos en balística y como balasto. Por ejemplo, puede reemplazar al plomo como material en plomadas para la pesca. Ha sido utilizado como substituto del plomo en munición de perdigones, balines y balas para dispersar multitudes. Los Países Bajos, Dinamarca, Inglaterra, Gales y Estados Unidos y numerosos otros países han prohibido el uso de perdigones de plomo para la caza de aves acuáticas, ya que muchas aves sufrían de envenenamiento por plomo al ingerir material al confundir los perdigones con piedrecillas que ingieren para mejorar el funcionamiento de su sistema digestivo, o incluso han prohibido el uso de plomo en todo tipo de caza como es el caso de los Países Bajos. En estos casos ciertas aleaciones de bismuto-estaño ofrecen una alternativa con propiedades similares al plomo para uso en balística. Sin embargo, dado que el bismuto es muy poco maleable, no es un material adecuado para fabricar balas de caza del tipo expansivas.
Al ser el bismuto un elemento denso con un peso atómico elevado, es utilizado para fabricar escudos de látex impregnados con bismuto para protección de los rayos-X durante exámenes médicos, tales como tomografías computerizadas con rayos X, principalmente porque se le considera un elemento no tóxico.12
La directiva de la Comunidad Europea sobre la restricción en cuanto al uso de substancias peligrosas que impulsa la reducción en cuanto al uso del plomo, ha ampliado el uso del bismuto en la industria electrónica como uno de los componentes de las soldaduras con bajo punto de fusión, reemplazando a las soldaduras tradicionales a base de plomo-estaño.13 Su baja toxicidad es especialmente importante para aquellas soldaduras que se utilizan en la fabricación de equipos para procesamiento de alimentos y tuberías de cobre para agua.14
Cosméticos y pigmentos[editar]
El oxicloruro de bismuto (BiOCl) a veces es utilizado en cosméticos, como pigmento en pintura para sombra de ojos, espray para el cabello y esmalte para uñas.151617 El compuesto se presenta en la naturaleza como el mineral bismoclita y la forma cristalina contiene capas de átomos que refractan la luz en forma cromática, produciendo un aspecto iridiscente similar al nácar de las perlas. Fue utilizado como cosmético en el antiguo Egipto y en muchas otras civilizaciones desde entonces. El término blanco de bismuto puede hacer referencia al oxicloruro de bismuto o al oxinitrato de bismuto (BiONO3), cuando son utilizados como pigmentos blancos.
Toxicidad[editar]
El salicilato de bismuto y el tioglicolato de bismuto utilizados para combatir la lúes y otros tipos de enfermedades infecciosas o parasitarias pueden causar, cuando se administran por vía parenteral, un cuadro de intoxicación por bismuto. El nitrato de bismuto administrado por vía oral puede metabolizarse en nitritos que se absorben en el intestino y son metahemoglobinizantes.18
La intoxicación aguda causa cefaleas, gastroenteritis, hepatopatías, anuria y shock. En la intoxicación crónica hay estomatitis, ribete gingival azulado, enteritis, ictericia, nefropatías y dermatitis de tipo exantemático o exfoliativo.18
El tratamiento de la intoxicación aguda consiste en administrar dimercaprol e ingerir abundante agua, salvo que haya daño renal, en cuyo caso debe tratarse de la misma forma que el hidroarsenisismo crónico regional endémico.18
Estados de oxidación[editar]
Bismuto (III)[editar]
El catión Bi3+, debido a su moderadamente alta acidez, se encuentra solamente en soluciones altamente ácidas en estado de equilibrio con sus óxidos. El catión Bi3+es incoloro debido a la estabilidad de su configuración electrónica ([Xe] 6s2) que impide transiciones electrónicas en longitudes de onda del espectro visible.19
Comportamiento ácido-base[editar]
Al aumentar la alcalinidad del medio, el catión nos Bi3+ forma principalmente las especies monohidroxobismuto(III), BiOH2+, y el catión bismutilo, BiO+.19
-
- Bi3+ + OH-
 Bi(OH)2+
Bi(OH)2+ - Bi3+ + 2OH- BiO+ + H2O
- Bi3+ + OH-
A pH altamente alcalino se produce el hidróxido de bismuto(III), Bi(OH)3, que por deshidratación genera el trióxido de dibismuto, Bi2O3, un sólido de color amarillo.19
-
- BiO+ + 4OH- Bi(OH)3↓ +H2O
- 2Bi(OH)3 (s) → Bi2O3 (s) + 3H2O
- BiO+ + 4OH-
-
- Bi2O3 (s) + 2OH- 2BiO2- + H2O
- Bi2O3 (s) + 2OH-
Presencia en compuestos orgánicos[editar]
Puede ser hallado formando uniones covalentes en ciertos compuestos orgánicos.



Bismuto (V)[editar]
A diferencia del bismuto (III), no se encuentra libre como catión ni en medios muy ácidos por su alta inestabilidad debida al alto valor de su relación carga/masa.19
Algunos de los compuestos en los que se encuentra son:
- Bi2O5, pentóxido de dibismuto
- BiF5, fluoruro de bismuto(V)
- NaBiO3, metabismutato de sodio
MOSCOVIO
El moscovio3 (anteriormente llamado unumpentio, Uup) es un elemento sintético de la tabla periódica cuyo símbolo es Mc y su número atómico es 115.4
Actualmente se conocen cuatro isótopos desde 287Mc hasta 290Mc. Se prevé que el isótopo más estable del moscovio sea el 299Mc, que contiene el número mágico de 184 neutrones. El isótopo con mayor número de neutrones conocido hasta la fecha es el 290Mc, con 175 neutrones. Es muy inestable, con una vida media de milésimas de segundo. Su nombre hace referencia a la provincia de Moscú, región a la que pertenece la ciudad rusa donde se descubrió, Dubná.

Descubrimiento[editar]
El 2 de febrero de 2004 se informó en la revista Physical Review C que un equipo integrado por científicos rusos en el Instituto Central de Investigaciones Nucleares en Dubná,5y los científicos estadounidenses en el Lawrence Livermore National Laboratory hicieron el descubrimiento del moscovio. El equipo informó que bombardearon americio 243 con calcio 48 para producir iones de cuatro átomos de moscovio. Estos átomos se desintegraron por emisión de partículas alfa en nihonio en aproximadamente 100 milisegundos.657 En agosto de 2013 otro experimento independiente confirmó el hallazgo del elemento.8
Otros elementos químicos sintetizados con anterioridad, como los de números atómicos 111 (roentgenio) y 112 (copernicio), tienen una existencia muy breve, de apenas milésimas de segundo, antes de desintegrarse. Esta característica es muy común entre los elementos transuránicos (los que aparecen en la tabla periódica más allá del uranio, cuyo número atómico es 92). Pero, cuando en 1999 se sintetizó el elemento 1149 (flerovio), se comprobó que es mucho más estable de lo que se pensaba: su vida media es de treinta segundos. A raíz de esto, muchos científicos pensaron que estaban a punto de encontrar la isla de estabilidad,10es decir, átomos superpesados pero estables durante años. El premio Nobel Glenn Seaborg predijo esta posibilidad en 1991.11 Calculó que se podría conseguir con algún isótopo de los elementos 114 o 115.1213
La clave de la estabilidad radica en que el núcleo del átomo sea lo más esférico posible, algo que, según Seaborg, puede ocurrir si posee al menos 298 nucleones (la suma de los protones y los neutrones). En el caso del experimento realizado recientemente por investigadores suizos, dirigidos por el doctor Heinz Gäggeler, la vida del nuevo átomo fue muy breve: una décima de segundo.14 Pero eso sólo indica que con el proceso empleado (bombardear un disco de americio con un rayo de iones de calcio) se ha obtenido un isótopo del elemento 115 que no llega a alcanzar la tan esquiva estabilidad.14 En el centro de investigación nuclear de Dubná(Rusia), donde se ha sintetizado el moscovio, varios equipos internacionales llevan años tratando de obtener nuevos elementos químicos.14 Allí se descubrió también el elemento 114 y, es muy probable que sea en este centro donde se consiga un isótopo estable de este elemento, aunque expertos astrónomos creen podría conseguirse en estado natural en el universo al igual que otros elementos superpesados.
Por otra parte y aunque parezca sorprendente, ésta no fue la primera vez que se tiene conocimiento de dicho elemento, ya que en noviembre de 1989, Bob Lazarapareció en una entrevista especial con el periodista e investigador George Knapp en la cadena de televisión de Las Vegas KLAS-TV para hablar de diversos asuntos, de los cuales, uno de ellos trataba acerca del descubrimiento y el uso en bases militares estadounidenses del elemento 115, también conocido como Moscovio.14
Nucleosíntesis[editar]
| Objetivo | Proyectil | CN | Resultado |
|---|---|---|---|
| 208Pb | 75As | 283Mc | Reacción aún no se ha intentado |
| 232Th | 55Mn | 287Mc | Reacción aún no se ha intentado |
| 238U | 51V | 289Mc | |
| 237Np | 50Ti | 287Mc | Reacción aún no se ha intentado |
| 244Pu | 45Sc | 289Mc | Reacción aún no se ha intentado |
| 243Am | 48Ca | 291Mc1516 | |
| 241Am | 48Ca | 289Mc | Reacción exitosa |
| 248Cm | 41K | 289Mc | Reacción aún no se ha intentado |
| 249Bk | 40Ar | 289Mc | Reacción aún no se ha intentado |
| 249Cf | 37Cl | 286Mc | Reacción aún no se ha intentado |
El moscovio en la tabla periódica de los elementos[editar]
El 5 de diciembre del 2016 la Unión Internacional de Química Pura y Aplicada (IUPAC) y la Unión Internacional de Física Pura y Aplicada (IUPAP) aprobaron su denominación junto a otros tres elementos como el nihonio, tenesino y oganesón. Además fue agregado a la tabla periódica de los elementos, al igual que los otros tres

GRUPO 6A DE LA TABLA PERIÓDICA
El grupo de los anfígenos o calcógenos es también llamado familia del oxígeno y es el grupo conocido antiguamente como VI A, y actualmente el grupo 16 (según la IUPAC) en la tabla periódica de los elementos, formado por los siguientes elementos: oxígeno (O), azufre (S), selenio (Se), telurio(Te), polonio (Po) y livermorio (Lv). El nombre de anfígeno en español deriva de la propiedad de algunos de sus elementos de formar compuestos con carácter ácido o básico.
Aunque todos ellos tienen seis electrones de valencia (última capa s2p4),1 sus propiedades varían de no metálicas a metálicas en cierto grado, conforme aumenta su número atómico.
El oxígeno y el azufre se utilizan abiertamente en la industria y el telurio y el selenio en la fabricación de semiconductores

OXIGENO
El oxígeno es un elemento químico de número atómico 8 y representado por el símbolo O. Su nombre proviene de las raíces griegas ὀξύς (oxys) («ácido», literalmente «punzante», en referencia al sabor de los ácidos) y –γόνος (-gonos) («productor», literalmente «engendrador»; es decir, "productor de ácidos"1), porque en la época en que se le dio esta denominación se creía, incorrectamente, que todos los ácidos requerían oxígeno para su composición. En condiciones normales de presión y temperatura, dos átomos del elemento se enlazan para formar el dioxígeno, un gas diatómicoincoloro, inodoro e insípido con fórmula O2. Esta sustancia constituye una importante parte de la atmósfera y resulta necesaria para sostener la vida terrestre.
Forma parte del grupo de los anfígenos en la tabla periódica y es un elemento no metálico altamente reactivo que forma fácilmente compuestos (especialmente óxidos) con la mayoría de elementos, excepto con los gases nobles helio y neón. Asimismo, es un fuerte agente oxidante y tiene la segunda electronegatividad más alta de todos los elementos, solo superada por el flúor.2 Medido por su masa, el oxígeno es el tercer elemento más abundante del universo, tras el hidrógeno y el helio,3 y el más abundante en la corteza terrestre ya que forma, prácticamente, la mitad de su masa.4 Debido a su reactividad química, no puede permanecer en la atmósfera terrestre como elemento libre sin ser reabastecido constantemente por la acción fotosintética de los organismos que utilizan la energía solar para producir oxígeno elemental a partir del agua. El oxígeno elemental O2 solamente empezó a acumularse en la atmósfera después de la aparición de estos organismos, aproximadamente hace 2500 millones de años.5 El oxígeno diatómico constituye el 20,8 % del volumen de la atmósfera terrestre.6
Dado que constituye la mayor parte de la masa del agua, es también el componente mayoritario de la masa de los seres vivos. Muchas de las moléculas más importantes que forman parte de los seres vivos, como las proteínas, los ácidos nucleicos, los carbohidratos y los lípidos, contienen oxígeno, así como los principales compuestos inorgánicos que forman los caparazones, dientes y huesos animales. El oxígeno elemental se produce por cianobacterias, algas y plantas y todas las formas complejas de vida lo usan para su respiración celular. Resulta tóxico para los organismos de tipo anaerobio obligado, las formas tempranas de vida que predominaban en la Tierra hasta que el O2 comenzó a acumularse en la atmósfera. Otra forma (alótropa) del oxígeno, el ozono (O3), ayuda a proteger la biosfera de la radiación ultravioleta a gran altitud, en la llamada capa de ozono, pero es contaminante cerca de la superficie, donde es un subproducto del esmog. A altitudes aún mayores de la órbita baja terrestre, el oxígeno atómico tiene una presencia significativa y causa erosión en las naves espaciales.7
Carl Wilhelm Scheele descubrió el oxígeno de forma independiente en Upsala en 1773, o incluso antes, y Joseph Priestley, en Wiltshire en 1774, pero el honor suele adjudicársele a Priestley debido a que publicó su trabajo antes. Antoine Lavoisier, cuyas investigaciones ayudaron a desacreditar la entonces popular teoría del flogisto de combustión y corrosión, acuñó el nombre «oxígeno» en 1777.8 Este se produce industrialmente mediante la destilación fraccionadade aire licuado, el uso de zeolita con ciclos de presión para concentrar el oxígeno del aire, la electrólisis del agua y otros medios. El oxígeno se utiliza en la producción de acero, plásticos y textiles; los propulsores de cohetes; la oxigenoterapia; y la asistencia para la respiración en aeronaves, submarinos, vuelos espaciales y submarinismoCaracterísticas[editar]
Estructura


En condiciones normales de presión y temperatura, el oxígeno es un gas incoloro e inodoro con fórmula molecular O2, en el que dos átomos de oxígeno se enlazan con una configuración electrónica en estado triplete. Este enlace tiene un orden de enlace de dos y se suele simplificar en las descripciones como un enlace doble9 o como una combinación de un enlace de dos electrones y dos enlaces de tres electrones.10
El oxígeno triplete —no debe confundirse con el ozono, O3— es el estado fundamental de la molécula O2,11 que cuenta con dos electrones desparejados que ocupan dos orbitales moleculares degenerados.nota 1 Estos orbitales se clasifican como antienlaces —debilitan el orden de enlace de tres a dos—, de manera que el enlace del dioxígeno es más débil que el triple enlace del nitrógeno diatómico, en el que todos los orbitales de los enlaces moleculares se rellenan, pero algunos orbitales de antienlace no lo están.11
En su forma normal de triplete, las moléculas de O2 son paramagnéticas; es decir, que en presencia de un campo magnéticoforman un imán, debido al momento magnético del espín de los electrones desparejados en la molécula y la interacción de canje negativa entre moléculas de O2 contiguas.12 Un imán atrae al oxígeno líquido hasta tal punto que, en demostraciones de laboratorio, un hilo de oxígeno líquido puede sostenerse contra su propio peso entre los polos de un imán potente.13nota 2
El oxígeno molecular singlete es un nombre dado a varias especies de O2 de mayor energía, en las que todos los espínes de los electrones se emparejan. Es mucho más reactivo con moléculas orgánicas habituales que el oxígeno molecular en sí mismo. En la naturaleza, el oxígeno singlete se suele formar con el agua en la fotosíntesis, usando la energía solar.15 También se produce en la troposfera a causa de la fotolisis del ozono por la luz de onda corta,16 así como por el sistema inmunitario como una fuente de oxígeno activo.17 En los organismos fotosintéticos —y posiblemente también en los animales—, los carotenoides juegan un papel fundamental en la absorción de energía del oxígeno singlete y la conversión de este a su estado no excitado antes de que pueda causar daño a los tejidos.18
Alótropos[editar]

El alótropo más normal del oxígeno elemental es el llamado dioxígeno (O2), que tiene una longitud de enlace de 121 pm y una energía de enlace de 498 kJ•mol−1.20 Esta es la forma que usan las formas de vida complejas, como los animales, en su respiración celular (véase rol biológico) y es la forma que tiene una gran importancia en la composición de la atmósfera terrestre (véase Abundancia).
El trioxígeno (O3) se conoce habitualmente como ozono y es un alótropo muy reactivo, dañino para el tejido pulmonar.21 El ozono se produce en la atmósfera superior cuando el O2 se combina con el oxígeno atómico a causa de la división del O2 por la radiación ultravioleta.8 Ya que el ozono es un poderoso absorbente en la región ultravioleta del espectro electromagnético, la capa de ozono de la atmósfera superior funciona como un escudo protector de la radiación que recibe el planeta.8 Cerca de la superficie terrestre, no obstante, es un contaminante formado como subproducto de las emisiones de automóviles.21 La molécula metaestable del tetraoxígeno (O4) no fue descubierta hasta 2001,2223 y se dio por descontado que existía en una de las seis fases del oxígeno sólido. En 2006 se demostró que esta fase, creada mediante la presurización del O2 a 20 GPa, es, de hecho, un clústernota 3 O8 de sistema trigonal.24 Este clúster tiene potencial para ser un oxidante mucho más potente que el O2 y el O3 y podría, por tanto, ser usado como propulsor de cohetes.2223 En 1990 se descubrió una fase metálica cuando el oxígeno sólido se somete a una presión superior a 96 GPa25 y se demostró en 1998 que a temperaturas muy bajas se convierte en superconductor.26
Propiedades físicas[editar]

El oxígeno es más soluble en agua que el nitrógeno; esta contiene aproximadamente una molécula de O2 por cada dos moléculas de N2,27 comparado con la proporción en la atmósfera, que viene a ser de 1:4. La solubilidad del oxígeno en el agua depende de la temperatura, disolviéndose alrededor del doble (14,6 mg•L−1) a 0 °C que a 20 °C (7,6 mg•L−1).1228 A 25 °C y 1 atmósfera de presión, el agua dulce contiene alrededor de 6,04 mililitros (ml) de oxígeno por litro, mientras que el agua marina contiene alrededor de 4,95 ml por litro.29 A 5 °C la solubilidad se incrementa hasta 9,0 ml (un 50 % más que a 25 °C) por litro en el agua y 7,2 ml (45 % más) en el agua de mar.
El oxígeno se condensa a 90,20 K (−182,95 °C, −297,31 °F) y se congela a 54,36 K (−218,79 °C, −361,82 °F).30 Tanto el O2líquido como el sólido son sustancias con un suave color azul cielo causado por la absorción en el rojo, en contraste con el color azul del cielo, que se debe a la dispersión de Rayleigh de la luz azul. El O2 líquido de gran pureza se suele obtener a través de la destilación fraccionada de aire licuado.31 El oxígeno líquido también puede producirse por condensación del aire, usando nitrógeno líquido como refrigerante. Es una sustancia altamente reactiva y debe separarse de materiales inflamables.32
Isótopos y origen estelar[editar]
El oxígeno que encontramos en la naturaleza se compone de tres isótopos estables: 16O, 17O y 18O, de los que el 16O es el más abundante (99,762 % de abundancia natural).33
La mayor parte del 16O se sintetiza al final del proceso de combustión del helio en una estrella masiva, pero otra parte se produce en el proceso de combustión del neón.34 El 17O surge fundamentalmente por la combustión del hidrógeno en helio durante el ciclo CNO, convirtiéndolo en un isótopo común en las zonas de combustión de hidrógeno en las estrellas.34 Por su parte, la mayoría del 18O se produce cuando el 14N —que abunda debido a la combustión CNO— captura un núcleo de 4He, lo que origina una gran abundancia de 18O en las zonas ricas en helio de las estrellas masivas.34
Se han caracterizado catorce radioisótopos, de los que los más estables son el 15O con un periodo de semidesintegración de 70,606 segundos.33 Todos los restantes isótopos radiactivos tienen periodos de semidesintegración inferiores a 27 segundos y la mayor parte de estos, inferiores a 83 milisegundos.33 La forma de descomposición de los isótopos más ligeros que el 16O es la descomposición β+353637 para producir nitrógeno y, para los más pesados que el 18O, la desintegración beta para formar flúor.33
Abundancia[editar]

El oxígeno es el elemento químico más abundante, por masa, en la biosfera, el aire, el mar y el suelo terrestres. Es, asimismo, el tercero más abundante en el universo, tras el hidrógeno y el helio.3 Alrededor del 0,9 % de la masa del Sol es oxígeno,6 que constituye también el 49,2 % de la masa de la corteza terrestre4 y es el principal componente de los océanos de la Tierra (88,8 % de su masa total).6 El oxígeno gaseoso es el segundo componente más abundante en la atmósfera terrestre, ya que supone un 20,8 % de su volumen y el 23,1 % de su masa (unas 1015 toneladas).638nota 4 La Tierra es una excepción entre los planetas del Sistema Solar por la alta concentración de oxígeno gaseoso en su atmósfera; por ejemplo, Marte (con un 0,1 % de O2 del total de su volumen) y Venus tienen concentraciones mucho menores. Sin embargo, el O2 que rodea a estos planetas proviene exclusivamente de la reacción que sufren moléculas que contienen oxígeno, como el dióxido de carbono, por efecto de la radiación ultravioleta.
La inusualmente alta concentración de oxígeno gaseoso en la Tierra es el resultado del ciclo de circulación. Este ciclo biogeoquímico describe el movimiento del oxígeno en el interior de sus tres principales reservas en el planeta: la atmósfera, la biosfera y la litosfera. El factor de conducción más importante en este ciclo es la fotosíntesis, responsable de la atmósfera moderna de la Tierra, que libera oxígeno en la atmósfera, mientras que los procesos de respiración y descomposición lo eliminan. En el equilibrio actual, la producción y el consumo tienen lugar con un ratio aproximado de 1/2000 de la totalidad del oxígeno atmosférico por año.
| Z | Elemento | Fracción másica en partes por millón |
|---|---|---|
| 1 | hidrógeno | 739 000 |
| 2 | helio | 240 000 |
| 8 | Oxígeno | 10 400 |
| 6 | Carbono | 4600 |
| 10 | neón | 1340 |
| 26 | Hierro | 1090 |
| 7 | nitrógeno | 960 |
| 14 | Silicio | 650 |
| 12 | Magnesio | 580 |
| 16 | Azufre | 440 |
El oxígeno no combinado también se da en soluciones en las masas de agua del planeta. La mayor solubilidad del O2 a baja temperatura (véase Propiedades físicas) tiene implicaciones importantes para la vida marina, ya que los océanos polares sostienen una densidad de vida mucho mayor debido a su superior contenido de oxígeno.nota 5 La cantidad de O2 en el agua puede haberse visto reducida por la contaminación hídrica, debido a la acción de la descomposición de las algas y otros biomateriales por un proceso llamado eutrofización. Los científicos evalúan este aspecto de la calidad del agua a través de la medición de su demanda biológica de oxígeno, o cantidad de O2 necesaria para restaurarla a una concentración normal.40
Rol biológico[editar]
Fotosíntesis y respiración[editar]

El oxígeno es liberado por las bacterias fotosintéticas, las algas y las plantas mediante la fotosíntesis. En el proceso inverso los organismos aerobios, mediante la respiración, usan el oxígeno para convertir los nutrientes en energía (ATP). La disminución de oxígeno provoca hipoxemia y su falta total, anoxia, lo que puede provocar la muerte del organismo.
En la naturaleza el oxígeno no combinado se produce por la fotodescomposición del agua durante la fotosíntesis. Según algunas estimaciones, las algas verdes y las cianobacterias de ambientes marinos proporcionan alrededor del 70 % del producido en la Tierra, y las plantas terrestres, el resto.41 Unos investigadores estiman que la contribución oceánica al oxígeno atmosférico es aún mayor, mientras que otros la sitúan por debajo, en torno a un 45 % del oxígeno atmosférico total del planeta cada año.42
Una fórmula global simplificada de la fotosíntesis es:43
La evolución fotolítica del oxígeno tiene lugar en las membranas tilacoides de los organismos fotosintéticos y requiere la energía de cuatro fotones.nota 6 Están implicados muchos procesos, pero el resultado es la formación de un gradiente de un protón a través de la membrana tilacoide, que se usa para sintetizar adenosín trifosfato (ATP) por la fotofosforilación.44 El O2restante tras la oxidación de la molécula de agua se libera a la atmósfera.nota 7
El dioxígeno molecular es esencial para la respiración celular en todos los organismos aerobios, ya que las mitocondrias lo usan para ayudar a generar adenosín trifosfato durante la fosforilación oxidativa. La reacción para la respiración aerobia es básicamente lo contrario que la fotosíntesis y se simplifica de la siguiente forma:

AZUFRE
El azufre es un elemento químico de número atómico 16 y símbolo S (del latín sulphur). Es un no metal abundante con un color amarillo característico. Dicho elemento es generado en estrellas masivas en las que predominan temperaturas que provocan la fusión entre un núcleo de silicio y otro de helio en un proceso denominado nucleosíntesis de supernovas.1
El azufre se encuentra en forma nativa en regiones volcánicas y en sus formas reducidas formando sulfuros y sulfosaleso bien en sus formas oxidadas como sulfatos. Es un elemento químico esencial constituyente de los aminoácidoscisteina y metionina y, por consiguiente, necesario para la síntesis de proteínas presentes en todos los organismos vivos. Se usa principalmente como fertilizante pero también en la fabricación de pólvora, laxantes, fósforos e insecticidas.

Características principales[editar]
Este no metal tiene un color amarillento fuerte, amarronado o anaranjado y arde con llama de color azul, desprendiendo dióxido de azufre. Es insoluble en agua pero se disuelve en disulfuro de carbono y benceno. Es multivalente, y son comunes los estados de oxidación -2, +2, +4, +6.
En todos los estados (sólido, líquido y gaseoso): según los químicos presenta formas alotrópicas cuyas relaciones no son completamente conocidas. Las estructuras cristalinas más comunes son el octaedro ortorrómbico (azufre α) y el prisma monoclínico (azufre β), siendo la temperatura de transición de una a otra de 96 °C; en ambos casos el azufre se encuentra formando moléculas de S8 con forma de anillo, y es la diferente disposición de estas moléculas la que provoca las distintas estructuras cristalinas. A temperatura ambiente, la transformación del azufre monoclínico en ortorrómbico, es más estable y muy lenta.
Al fundir el azufre, se obtiene un líquido que fluye con facilidad formado por moléculas de S8. Sin embargo, si se calienta, el color se torna marrón algo rojizo, y se incrementa la viscosidad. Este comportamiento se debe a la ruptura de los anillos y la formación de largas cadenas de átomos de azufre, que pueden alcanzar varios miles de átomos de longitud, que se enredan entre sí disminuyendo la fluidez del líquido; el máximo de la viscosidad se alcanza en torno a los 200 °C. Enfriando rápidamente este líquido viscoso se obtiene una masa elástica, de consistencia similar a la de la goma, denominada «azufre plástico» (azufre γ) formada por cadenas que no han tenido tiempo de reordenarse para formar moléculas de S8; transcurrido cierto tiempo la masa pierde su elasticidad cristalizando en el sistema rómbico. Estudios realizados con rayos X muestran que esta forma deforme puede estar constituida por moléculas de S8 con estructura de hélice espiral.
En estado vapor también forma moléculas de S8, pero a 780 °C ya se alcanza el equilibrio con moléculas diatómicas y por encima de aproximadamente 1800 °C la disociación es completa y se encuentran átomos de azufre.
Además de en trozos, barras o polvo grueso, existe en el mercado una presentación en forma de polvo muy fino, llamada "Flor de azufre", que puede obtenerse por precipitación en medio líquido o por sublimación de su vapor sobre una placa metálica fría. Estas son sus reacciones: S+Zn=ZnS 2Al+3S=Al²S³ S+O²=SO² 6S+HNO³=H²SO⁴+6NO²+2H²O

Aplicaciones[editar]
El azufre se usa en multitud de procesos industriales, como la producción de ácido sulfúrico para baterías, la fabricación de pólvora y el vulcanizado del caucho.
Los sulfitos se usan para blanquear el papel y en fósforos. El tiosulfato de sodio o amonio se emplea en la industria fotográfica como «fijador» ya que disuelve el bromuro de plata; y el sulfato de magnesio (sal de Epsom) tiene usos diversos como laxante, exfoliante, o suplemento nutritivo para plantas.
También el azufre se emplea en la industria enológica como antiséptico. Uno de sus principales usos es como anhídrido sulfuroso.
Historia[editar]
El azufre es conocido desde la Antigüedad, y ya los egipcios lo utilizaban para purificar los templos.
En el Génesis (19,24), los hebreos decían que Dios (Yahvé) hizo llover sobre Sodoma y Gomorra azufre y fuego desde el cielo.
Homero recomendaba, en el siglo IX a. C., evitar la pestilencia mediante la quema de azufre (zeio en griego, relacionado con zeos-Zeus).
Según el Diccionario sánscrito-inglés (1899) de Monier Monier-Williams, en sánscrito al azufre se lo llamaba śulbāri(pronunciado /shulbári/), siendo śulba o śulva: ‘cobre’, y a-rí o a-rís: ‘enemigo, envidioso’ (lit. ‘no liberal’).
En Apocalipsis 20:10 se dice que el diablo será lanzado a un lago de fuego y azufre.
Durante toda la Edad Media se vinculó a Satanás con los olores sulfurosos (relacionados con los volcanes, que se suponían eran entradas a los infiernossubterráneos).
Abundancia y obtención[editar]

El azufre es un elemento muy abundante en la corteza terrestre, se encuentra en grandes cantidades combinado en forma de sulfuros (pirita, galena) y de sulfatos (yeso). En forma nativa se encuentra en las cercanías de aguas termales, zonas volcánicas y en minas de cinabrio, galena, esfalerita y estibina, y en Luisiana (Estados Unidos, primer productor mundial) se extrae mediante el proceso Frasch consistente en inyectar vapor de agua sobrecalentado para fundir el azufre que posteriormente es bombeado al exterior utilizando aire comprimido.También se obtiene separándolo del gas natural, si bien su obtención anteriormente era a partir de depósitos de azufre puro impregnado en cenizas volcánicas (Italia, y más recientemente Argentina).
También está presente, en pequeñas cantidades, en combustibles fósiles (carbón y petróleo) cuya combustión produce dióxido de azufre que combinado con agua produce la lluvia ácida; para evitarlo las legislaciones de los países industrializados exigen la reducción del contenido de azufre de los combustibles, constituyendo este azufre, posteriormente refinado, un porcentaje importante del total producido en el mundo. También se extrae del gas natural que contiene sulfuro de hidrógeno que una vez separado se quema para obtener azufre:
- 2 H2S + O2 → 2 S + 2 H2O
El color distintivo de Ío, la luna volcánica de Júpiter, se debe a la presencia de diferentes formas de azufre en estado líquido, sólido y gaseoso. El azufre se encuentra, además, en varios tipos de meteoritos, y se cree que la mancha oscura que puede observarse cerca del cráter lunar Aristarco puede ser un depósito de azufre.
Estados de oxidación y Compuestos[editar]
La variedad de compuestos azufrados son consecuencia de una gran variedad de posibles estados de oxidación del átomo de azufre. En la Tabla2 se recojen algunos ejemplos de familias de compuestos azufrados, en función del estado de oxidación del azufre.
Muchos de los olores desagradables de la materia orgánica se deben a compuestos de la materia que contienen azufre como el sulfuro de hidrógeno. Disuelto en agua es ácido (pKa1 = 7,00, pKa2 = 12,92) y reacciona con los metales. Los sulfuros metálicos se encuentran en la naturaleza, sobre todo el de hierro (pirita) que puede presentar resistencia negativa y la galena, sulfuro de plomo que es un semiconductor natural que fue usado como rectificador.
El nitruro de azufre polímero (SN)x, sintetizado en 1975 por Alan G. MacDiarmid y Alan J. Heeger, presenta propiedades metálicas, a pesar de estar constituido por no metales, e inusuales propiedades eléctricas y ópticas. Este trabajo sirvió de base para el posterior desarrollo, con Hideki Shirakawa, de plásticos conductores y semiconductores que motivó la concesión del Nobel de Química, en 2000, a los tres investigadores.
Los óxidos más importantes son el dióxido de azufre, SO2 (formado por la combustión del azufre) que en agua forma una solución de ácido sulfuroso, y el trióxido de azufre, SO3, que en solución forma el ácido sulfúrico; siendo los sulfitos y sulfatos las sales respectivas.

SELENIO
El selenio es un elemento químico de la tabla periódica cuyo símbolo es Se, cuyo número atómico es 34. Pertenece a la familia de los no metales.

Características principales[editar]
El selenio se puede encontrar en varias formas alotrópicas. El selenio amorfo existe en tres formas, la vítrea, negra, obtenida al enfriar rápidamente el selenio líquido, funde a 180 °C y tiene una densidad de 4,28 g/cm3; la roja, coloidal, se obtiene en reacciones de reducción; el selenio gris cristalino de estructura hexagonal, la forma más común, funde a 220,5 °C y tiene una densidad de 4,81 g/cm3; y la forma roja, de estructura monoclínica, funde a 221 °C y tiene una densidad de 4,39 g/cm3.
Presenta el efecto fotoeléctrico, convirtiendo la luz en electricidad, y, además, su conductividad eléctrica aumenta al exponerlo a la luz. Por debajo de su punto de fusión es un material semiconductor tipo p, y se encuentra en su forma natural.
Aplicaciones[editar]
El selenio se usa con diversos fines. Su derivado, el selenio de amonio, por ejemplo, se ocupa en la fabricación de vidrio.[cita requerida] Otro derivado, el sulfuro de selenio, se usa en lociones y champúes como tratamiento para la dermatitis seborreica.3
Papel biológico[editar]
El selenio es un micronutriente para todas las formas de vida conocidas que se encuentra en el pan, los cereales, el pescado, las carnes, las lentejas, la cáscara de las patatas y los huevos. Está presente en el aminoácido selenocisteínay también se puede encontrar como selenometionina, reemplazando al azufre de la cisteína y la metioninarespectivamente. Forma parte de las enzimas glutatión peroxidasa y tiorredoxina reductasa.4
Es antioxidante, ayuda a neutralizar los radicales libres, induce la apoptosis, estimula el sistema inmunológico e interviene en el funcionamiento de la glándula tiroides. Las investigaciones realizadas sugieren la existencia de una correlación entre el consumo de suplementos de selenio y la prevención del cáncer en humanos.4 De manera similar, algunos estudios han comprobado que algunas concentraciones de selenio resultan quimioprotectoras frente a la apoptosis inducida por estrés oxidativo.5 Aún es tema de investigación, pero se sabe que la forma química en la que se encuentra el selenio (selenito, selenato o selenoaminoácidos) afecta a su absorción y a su posible toxicidad. Los datos actuales apuntan a que la forma orgánica (formando parte de proteínas como selenoaminoácidos) es la más beneficiosa para los animales. Además potencia el buen humor.[cita requerida]
La deficiencia de selenio es relativamente rara, pero puede darse en pacientes con disfunciones intestinales severas o con nutrición exclusivamente parenteral, así como en poblaciones que dependan de alimentos cultivados en suelos pobres en selenio. La ingesta diaria recomendada para adultos es de 55-70 μg; más de 400 μg puede provocar efectos tóxicos (selenosis).
Historia[editar]
El selenio (del griego σελήνιον,"selénion", resplandor de la Luna y por selene o artemisa la diosa griega de la luna y los animales) fue descubierto en 1817 por Jöns Jacob Berzelius. Al visitar la fábrica de ácido sulfúrico de Gripsholm observó un líquido pardo rojizo que calentado al soplete desprendía un olor fétido que se consideraba entonces característico y exclusivo del telurio —de hecho su nombre deriva de su relación con este elemento ya que telurio proviene del latín Tellus, la Tierra— resultando de sus investigaciones el descubrimiento del selenio. Más tarde, el perfeccionamiento de las técnicas de análisis permitió detectar su presencia en distintos minerales pero siempre en cantidades extraordinariamente pequeñas.
Abundancia y obtención[editar]
El selenio se encuentra muy distribuido en la corteza terrestre en la mayoría de las rocas y suelos se halla en concentraciones entre 0,1 y 2,0 ppm. Raramente se encuentra en estado nativo obteniéndose principalmente como subproducto de la refinación del cobre ya que aparece en los lodos de electrólisis junto al telurio (5-25 % Se, 2-10 % Te). La producción comercial se realiza por tostación con cenizas de sosa o ácido sulfúrico de los lodos.
Primeramente se añade un aglomerante de cenizas de sosa y agua a los lodos para formar una pasta dura que se extruye o corta en pastillas para proceder a su secado. La pasta se tuesta a 530-650 °C y se sumerge en agua resultando selenio hexavalente que se disuelve como selenato de sodio (Na2SeO4). Este se reduce a seleniuro de sodio calentándolo de forma controlada obteniendo una solución de un vivo color rojo. Inyectando aire en la solución el seleniuro se oxida rápidamente obteniéndose el selenio. La reducción del selenio hexavalente también puede hacerse empleando ácido clorhídrico concentrado, o sales ferrosas e iones cloro como catalizadores.
El segundo método consiste en mezclar los lodos de cobre con ácido sulfúrico tostando la pasta resultante a 500-600 °C para obtener dióxido de selenio que rápidamente se volatiliza a la temperatura del proceso. Este se reduce a selenio elemental durante el proceso de lavado con dióxido de azufre y agua, pudiendo refinarse posteriormente hasta alcanzar purezas de 99,5-99,7 % de selenio.
Los recursos de selenio asociados a los depósitos de cobre identificados rondan las 170.000 toneladas y se estima que existen alrededor de 425 000 toneladas más en depósitos de cobre y otros metales aún no explotados. El carbón suele contener entre 0,5 y 12 ppm de selenio, es decir, unas 80 o 90 veces el promedio que se encuentra en las minas de cobre, sin embargo su recuperación no se prevé que pueda realizarse en un futuro próximo.
Isótopos[editar]
El selenio tiene seis isótopos naturales, cinco de los cuales son estables: 74Se, 76Se, 77Se, 78Se, y 80Se. Los tres últimos también se presentan como productos de fusión, junto con 79Se que tiene una vida media de 295 000 años.
Precauciones[editar]
El selenio está considerado un elemento peligroso para el medio ambiente por lo que sus compuestos deben almacenarse en áreas secas evitando filtraciones que contaminen las aguas. Los residuos de selenio se tratan en solución ácida con sulfito de sodio, calentándolo después para obtener el selenio elemental que presenta una menor biodisponibilidad.

TELURIO
El telurio o teluro2 es un elemento químico cuyo símbolo es Te y su número atómico es 52. Es un metaloide que se encuentra en el grupo 16 y el periodo 5 de la Tabla periódica de los elementos.
Fue descubierto en 1782 en minerales de oro por Franz-Joseph Müller von Reichenstein, inspector jefe de minas en Transilvania (Rumanía), denominándolo metallum problematicum. En principio se confundió el telurio con el antimonio. Fue Martin Heinrich Klaproth, en 1798, quien examinó el «metal problemático» de Müller y lo llamó telurio.3
El telurio es un elemento relativamente estable, insoluble en agua y ácido clorhídrico, pero soluble en ácido nítrico y en agua regia. Reacciona con un exceso de cloro para formar dicloruro de teluro, TeCl2 y tetracloruro de teluro, TeCl4. Se oxida con ácido nítrico y produce dióxido de teluro, TeO2, y con ácido crómico para dar ácido telúrico, H2TeO4. En combinación con el hidrógeno y ciertos metales, forma telururos, como el telururo de hidrógeno, H2Te, y el telururo de sodio, Na2Te. El teluro tiene un punto de fusión de 452° C, un punto de ebullición de 990° C, y una densidad relativa de 6,25. Su masa atómica es 127,60. [cita requerida]
Los compuestos de telurio se usan ampliamente en la química orgánica sintética para la reducción y oxidación, ciclofuncionalización, deshalogenación, reacciones de generación de carbaniones y eliminación de grupos protectores.4 Los compuestos organometálicos son intermedios en la síntesis de aminas, dioles y productos naturales.56 El telurio es un componente de importancia clave en los catalizadores de óxidos mixtos de alto rendimiento para la oxidación selectiva catalítica heterogénea de propano a ácido acrílico.78 En presencia de vapor de agua, la superficie del catalizador se enriquece en telurio y vanadio lo que se traduce en la mejora de la producción de ácido acrílico.910 El telurio puede usarse en sensores de amoníaco11 y cristales de telurita.12
Isótopos[editar]
Se conocen 29 isótopos del telurio, con masas atómicas que fluctúan entre 108 y 137. En la naturaleza hay 8 isótopos del telurio, de los cuales tres son radiactivos. El 128Te tiene el periodo de semidesintegración más largo conocido de todos los radioisótopos de telurio (2,2·1024 años). El telurio es el elemento con menor número atómico que puede experimentar la desintegración alfa. Con los isótopos del 106Te al 110Te, puede experimentar este tipo de desintegración.
Abundancia y obtención[editar]

El telurio puede obtenerse combinado con oro en la calaverita,
un mineral metálico relativamente poco abundante. [cita requerida]
En abril de 2017 se publicó el hallazgo del mayor yacimiento de telurio del mundo, en aguas de las Islas Canarias (España), en los montes submarinos situados dentro de las aguas canarias llamadas "las abuelas de Canarias" (Drago, Bimbache, Ico, Pelicar, Malpaso, Tortuga e Infinito y Las Abuelas). Se calcula que el yacimiento tiene un total de unas 2670 toneladas de Telurio, unas 50 000 veces más que el hallazgo más grande encontrado hasta ahora.1314
Telururo de cadmio
El telururo de cadmio (CdTe) es un compuesto cristalino formado por cadmio y telurio. Se utiliza como ventana ópticade infrarrojos y como material de célula solar.1 Por lo general se intercala con sulfuro de cadmio para formar una célula fotovoltaica de unión pn. Normalmente, las células de CdTe utilizan una estructura n-i-p .

POLONIO
El polonio es un elemento químico en la tabla periódica de los elementos cuyo símbolo es Po y su número atómicoes 84. Se trata de un raro metal altamente radiactivo, químicamente similar al telurio y al bismuto, presente en minerales de uranio.

Características[editar]
Esta sustancia se disuelve con mucha facilidad en ácidos, pero es sólo ligeramente soluble en alcalinos. Está químicamente relacionado con el teluro y el bismuto. El polonio es un metal volátil, reducible al 50% tras 45 horas al aire a una temperatura de 54,8 °C (328 K). Ninguno de los 50 isotopos [numero estimado] de polonio es estable. Es extremadamente tóxico y altamente radiactivo. Se ha encontrado polonio en minerales de uranio, humo de tabaco y como contaminante. Todos los elementos a partir del polonio son significativamente radiactivos. Se encuentra en el grupo 16 y su número atómico es 84.
Aplicaciones[editar]
Mezclado o aleado con berilio, el polonio puede ser una fuente de neutrones.
Se utiliza también en dispositivos destinados a la eliminación de carga estática, en cepillos especiales para eliminar el polvo acumulado en películas fotográficas y también en fuentes de calor para satélites artificiales o sondas espaciales.
Polonio-210[editar]
Este isótopo de polonio es un emisor alfa con un período de semidesintegración de 138,39 días. Un miligramo de 210Po emite tantas partículas alfa como 5 gramos de radio. Por ello libera gran cantidad de energía, alcanzando los dispositivos productores de calor (en los Generadores Termoeléctricos de Radioisótopos o RTG en inglés) una temperatura superior a los 750 K con tan sólo medio gramo. Un único gramo de este isótopo genera 130 vatios de potencia calórica.
El 210Po se ha utilizado como fuente ligera de calor para dar energía a las células termoeléctricas de algunos satélites artificiales y sondas lunares.
Polonio-210 en el tabaco[editar]
La presencia de polonio en el humo de tabaco se conoce desde principios de los años 60.12 Algunas de las empresas tabacaleras más importantes del mundo han investigado, sin éxito, formas de eliminar esta sustancia durante 40 años. Sin embargo, nunca publicaron los resultados.3
El polonio-210 contenido en los fertilizantes fosfatados es absorbido por las raíces de plantas (como el tabaco) y almacenado en sus tejidos.456 Las plantas fertilizadas con fosfatos de roca contienen polonio-210, y la radiación alfa que emite se estima que causa alrededor de 11 700 muertes anuales en todo el mundo por cáncer de pulmón.378
Efectos negativos[editar]
La ingesta o inhalación de una cantidad excesiva puede tener las mismas consecuencias que tuvo la radiación de Hiroshima, pero individualmente. Estos efectos se pueden notar a partir del tercer día, no son inmediatos. Los primeros síntomas son la caída del pelo y las molestias gastrointestinales. A continuación, el hígado y los riñones fallan; se paraliza el metabolismo y la médula ósea. La muerte será por fallo multiorgánico. Si la exposición es reducida sólo pueden aparecer síntomas gastrointestinales. Estos son los siguientes síntomas:
- Los efectos por exposición son fallos orgánicos, caída del cabello, dolores gástricos, daños de pulmón y riñones y fallo total del sistema inmune.
- si la intoxicación es baja los síntomas se atenúan hasta desaparecer en unos días.
- el tratamiento medico pasa por un quelante o compuesto químico que ayuda a expulsar el polonio 210 a través de las heces y la orina.
Historia[editar]
_(cropped).jpg)
También conocido como Radio F, el polonio fue descubierto por Marie Curie-Skłodowska y Pierre Curie en 1898, y fue posteriormente renombrado en honor a la tierra natal de Marie Curie, Polonia. En aquella época, Polonia no era un país independiente y se encontraba bajo el dominio de Rusia, Prusia y Austria, y Marie albergaba la esperanza de que este nombramiento le añadiría notoriedad. Fue el primer elemento cuyo nombre derivaba de una controversia política.
Fue el 1º elemento descubierto por el matrimonio Curie mientras investigaban las causas de la radiactividad de la pechblenda. La pechblenda, tras eliminar el uranio y el radio, era incluso más radiactiva que estos elementos juntos. Esto les llevó a encontrar el nuevo elemento. El electroscopio lo mostró separándolo con bismuto.
Obtención[editar]
Aunque es un elemento de procedencia natural, sólo está presente en los minerales de uranio natural en razón de 100 microgramos por tonelada.
En 1934 se demostró que, cuando el bismuto natural (209Bi) es bombardeado con neutrones, se crea 210Bi, que se transforma mediante una desintegración beta en Polonio-210. Se puede crear polonio en cantidades de miligramos mediante este procedimiento, utilizando flujos de neutrones grandes, como los que se encuentran en los reactores nucleares.
Precauciones[editar]
El polonio es un elemento altamente tóxico (DL50 = 10ng (inhalados) o 50 ng (ingeridos) en seres humanos), radiactivo y de peligroso manejo. Incluso en cantidades de microgramos, el manejo de 210Po, es muy peligroso y requiere de equipamiento especial utilizado bajo estrictos procedimientos de seguridad.
Ejemplo de Peligrosidad[editar]
En 2006 el ex espía ruso Aleksandr Litvinenko fue asesinado con 210Po, supuestamente debido a su investigación por el asesinato de la periodista Anna Politkóvskaya. Esto está reflejado en uno de los capítulos de la serie Mil maneras de morir, con una adaptación gráfica del envenenamiento del espía.
Alexander Litvinenko fue empleado por el Centro Nacional de Inteligencia (CNI) en una investigación sobre los posibles vínculos entre la mafia rusa y el presidente del país, Vladímir Putin. Según los diarios británicos Daily Mail y Evening Standar, esta revelación fue hecha por Ben Emmerson, abogado de la esposa del fallecido, durante la audiencia preliminar para la investigación de la muerte en Londres del antiguo agente del KGB, que fue envenenado con polonio 210 supuestamente vertido en una taza de té que bebió en el hotel Mayfair durante una reunión con antiguos compañeros de los servicios de seguridad.
Fuentes del CNI, rehusaron confirmar o desmentir esta revelación, alegando que la ley impide taxativamente decir si una persona, aunque esté muerta, fue o no una fuente, un miembro del servicio o un colaborador. No obstante, seis meses antes de morir envenenado, Litvinenko contactó con policías españoles para explicarles qué papel desempeñaban ciertos hombres de negocios involucrados con la mafia rusa. El ex agente ruso dio algunas pistas sobre la importancia que tenían algunos jefes mafiosos y qué tipo de relaciones podían mantener con altas instancias del Estado ruso.
Según una investigación de Al Jazeera de julio de 2012, también Yaser Arafat habría muerto envenenado con Polonio 210.

LIVERMORIO
El livermorio (anteriormente llamado ununhexio, Uuh) es el nombre del elemento sintético de la tabla periódica cuyo símbolo es Lv y su número atómico es 116.
Su nombre viene dado en honor al Laboratorio Nacional Lawrence Livermore (Lawrence Livermore National Laboratory), en Livermore, California.

Historia[editar]
En 1999, investigadores del Laboratorio Nacional Lawrence Berkeley anunciaron la creación del elemento 116, en un artículo publicado en una revista de los EEUU llamada Physical Review Letters, explican que lo hicieron cuando se observó el decaimiento-α de un átomo de mayor número atómico. El año siguiente publicaron su retracción tras ver que no eran capaces de volver a hacer el experimento.3 En junio de 2002, el director del laboratorio anunció que los datos del experimento habían sido falseados por su autor principal Victor Ninov.
En junio de 2000, el Instituto Conjunto para la Investigación Nuclear, en la ciudad de Dubna, realizó estudios por los cuales se describió el decaimiento-α del isótopo 292Uuh que se produjo en la reacción de fusión de un núcleo de 248 Cm al ser bombardeado con iones de 48 Ca acelerados por un ciclotrón, como producto secundario se obtuvieron 4 neutrones. Tiene una vida media de cerca de 6 milisegundos (0,006 segundos). Luego de esto tiene un decaimiento-α en 288Fl (Flerovio) seguido de dos más consecutivos en otros átomos de menor número atómico para más tarde tener una fisión espontánea.
Nuevos experimentos se hicieron entre finales de 2000 e inicios de 2001, pero estos fallaron en crear un nuevo átomo.
El 2 de mayo de 2001, el mismo instituto informó sobre la síntesis de un segundo átomo en su cuarta ronda de estudios, y que las propiedades confirmaron una región de la estabilidad "aumentada", aunque son acreditados de gran calidad y cuidadosos la confirmación de estos resultados está todavía pendiente por falta de estudios entre ellos un estudio con rayos X que pruebe la conexión entre las reacciones y descendientes.
En octubre de 2006 se anunció que, en tres ocasiones, bombardeando átomos de californio-249 con iones de calcio-48 producían ununoctio (elemento 118), que posteriormente decaía a ununhexio en tiempos de milisegundos.4 Confirmando esto, la síntesis del elemento 116 habrá sido demostrada definitivamente.
La reacción que crea el livermorio es:

Decae en 47 milisegundos a un isótopo previamente identificado del elemento 114, Fl.

Aplicaciones[editar]
Por su inestabilidad, vida media tan reducida y dificultad de obtención, en la actualidad son nulas las aplicaciones industriales, comerciales o propagandísticas de este elemento muy pesado por lo que su aplicación se relega sólo a la investigación científica.
El nombre definitivo del Livermorio[editar]
Ununhexio es un nombre temporal IUPAC. Algunos científicos del Joint Institute for Nuclear Research propusieron para este elemento el nombre de "Flyorovium" (Fl) - en honor a G. N. Flyorov, director del grupo que sintetizó los elementos del 102 al 110. Pero no hay ninguna mención que confirme este nombre para el elemento aún.
El 8 de diciembre de 2011, la división de Química Inorgánica de la IUPAC confirmó el nombre y símbolo de este elemento a la vez que el nombre y símbolo del Ununquandio, ahora llamado Flerovio (Fl). El nombre definifivo decidido fue Livermorio (Lv), en honor al Laboratorio Nacional de la ciudad de Livermore, California

GRUPO 7A DE LA TABLA PERIÓDICA
Los halógenos (del griego, formador de sales) son los elementos químicos que forman el grupo 17 (XVII A, utilizado anteriormente) o grupo VII A de la tabla periódica: flúor (F), cloro (Cl), bromo (Br), yodo (I), astato (At) y téneso (Ts). Este último también está en los metales del bloque f.
En estado natural se encuentran como moléculas diatómicas químicamente activas [X2]. Para llenar por completo su último nivel energético (s2p5) necesitan un electrón más, por lo que tienen tendencia a formar un ion mononegativo, X-. Este ion se denomina haluro; las sales que lo contienen se conocen como haluros. Poseen una electronegatividad ≥ 2.5 según la escala de Pauling, presentando el flúor la mayor electronegatividad, y disminuyendo ésta al bajar en el grupo. Son elementos oxidantes (disminuyendo esta característica al bajar en el grupo), y el flúor es capaz de llevar a la mayor parte de los elementos al mayor estado de oxidación.
Muchos compuestos orgánicos sintéticos, y algunos naturales, contienen halógenos; a estos compuestos se les llama compuestos halogenados. La hormona tiroideacontiene átomos de yodo. Los cloruros tienen un papel importante en el funcionamiento del cerebro mediante la acción del neurotransmisor inhibidor de la transmisión GABA (ácido gamma-amino butírico).
Algunos compuestos presentan propiedades similares a las de los halógenos, por lo que reciben el nombre de pseudohalógenos. Puede existir el pseudohalogenuro, pero no el pseudohalógeno correspondiente. Algunos pseudohalogenuros: cianuro (CN-), tiocianato (SCN-), fulminato (CNO-), etcétera.
Los fenicios y los griegos de la antigüedad utilizaron la sal común para la conservación de alimentos, especialmente en la salazón del pescado.
FLUOR
El flúor es el elemento químico de número atómico 9 situado en el grupo de los halógenos (grupo 17) de la tabla periódica de los elementos. Su símbolo es F.
Es un gas a temperatura ambiente, de color amarillo pálido, formado por moléculas diatómicas F2. Es el más electronegativo y reactivo de todos los elementos. En forma pura es altamente peligroso, causando graves quemaduras químicas al contacto con la piel.

Características principales[editar]
El flúor es el elemento más electronegativo y reactivo y forma compuestos con prácticamente todo el resto de elementos, incluyendo los gases nobles xenón y radón. Su símbolo es F. Incluso en ausencia de luz y a bajas temperaturas, el flúor reacciona explosivamente con el hidrógeno. El flúor diatómico, F2, en condiciones normales es un gas corrosivo de color amarillo casi blanco, fuertemente oxidante. Bajo un chorro de flúor en estado gaseoso, el vidrio, metales, agua y otras sustancias, se queman en una llama brillante. Siempre se encuentra en la naturaleza combinado y tiene tal afinidad por otros elementos, especialmente silicio, que no se puede guardar en recipientes de vidrio.
En disolución acuosa, el flúor se presenta normalmente en forma de ion fluoruro, F-. Otras formas son fluorocomplejos como el [FeF4]-, o el H2F+.
Los fluoruros son compuestos en los que el ion fluoruro se combina con algún resto cargado positivamente.
Aplicaciones[editar]
- El politetrafluoroetileno (PTFE), también denominado teflón, se obtiene a través de la polimerización de tetrafluoroetileno que a su vez es generado a partir de clorodifluorometano, que se obtiene finalmente a partir de la fluoración del correspondiente derivado halogenado con fluoruro de hidrógeno (HF).
- También a partir de HF se obtienen clorofluorocarburos (CFC), hidroclorofluorocarburos (HClFC) e hidrofluorocarburos (HFC).
- Se emplea flúor en la síntesis del hexafluoruro de uranio, UF6, es el gas más pesado conocido y se emplea en el enriquecimiento de uranio 235U.
- El fluoruro de hidrógeno se emplea en la obtención de criolita sintética, Na3AlF6, la cual se usa en el proceso de obtención de aluminio.
- Hay distintas sales de flúor con variadas aplicaciones[cita requerida]. El fluoruro de sodio, NaF, se emplea como agente fluorante; el difluoruro de amonio, NH4HF2, se emplea en el tratamiento de superficies, anodizado del aluminio, o en la industria del vidrio; el trifluoruro de boro, BF3, se emplea como catalizador; etc.
- Algunos fluoruros se añaden a las pastas de dientes para la prevención de caries (principalmente el fluoruro de sodio).
- En algunos países se añade fluoruro a las aguas potables para prevenir la aparición de caries, de lo que se suele avisar a la población. Algunos países como Estados Unidos o Australia fluoran el agua potable, mientras que otros como Alemania lo prohíben.[cita requerida]
- Se emplea flúor monoatómico en la fabricación de semiconductores.
- El hexafluoruro de azufre, SF6, es un gas dieléctrico con aplicaciones electrónicas. Este gas contribuye al efecto invernadero y está recogido en el Protocolo de Kioto.
Historia[editar]
A causa de ser tan reactivo y peligroso, el flúor no fue aislado hasta tiempos relativamente recientes, puesto que en estado puro es sumamente peligroso y es necesario manejarlo con extremo cuidado.1
El primer compuesto de flúor (del latín fluere, que significa "fluir") que se conoce data de los años 1500, en Alemania. Se trata de la fluorita (CaF2), por entonces llamada flúores, después espato de flúor. Es un mineral raro, que se funde fácilmente y era utilizado como fundente, para fundir otros minerales con mayor facilidad al mezclarlo con flúores. El mineralogista Georgius Agricola describió el mineral en 1529.1
En 1670, Enrique Schwandhard descubrió que al someter al mineral a algunos ácidos, desprendía un vapor muy corrosivo, que incluso corroía el vidrio. Utilizó esta propiedad para elaborar dibujos sobre el vidrio, por lo que mantuvo en secreto la forma de obtenerlo.2
Solo muy lentamente se avanzó en el estudio de este mineral. En 1768, Andrés Segismundo Sargraf estudió el mineral y obtuvo nuevamente el extraño vapor, informando sobre la característica que ataca al vidrio.2
Sin embargo, el primero en estudiar el gas fue Carlos Sabéele en 1780. A él se le atribuye el descubrimiento del ácido fluorhídrico. Murió a los 44 años, muy probablemente a causa de una intoxicación sistemática con los productos que manejaba.2
En 1813, Ampère hizo la hipótesis de que el ácido fluorhídrico era un compuesto de hidrógeno con un elemento todavía no descubierto. Esta hipótesis la hizo por la analogía que tiene este ácido con el muriático, del que se descubrió el cloro apenas tres años antes. Comunicó su hipótesis a Humphry Davy. Ampère sugirió el nombre de "pthor" al nuevo elemento, pero Davy se inclinó por el nombre "flúor".34
Desde entonces se sucedió una serie de intentos de aislar el flúor, todos fallidos, y la mayoría con accidentes de intoxicación. Comenzó el mismo Davy por medio de electrólisis, descomponiendo el fluoruro cálcico, pero no lo logró debido a que una vez aislado el flúor en el electrodo positivo, se combinaba rápidamente con cualquier elemento que estuviese cerca. En el proceso se intoxicó y probablemente a causa de eso tuvo una muerte temprana.5
En 1830 los hermanos Tomás y Jorge Knox intentaron aislar el flúor por medios químicos usando cloro. No lo lograron y también se intoxicaron seriamente.5
P. Louyel también lo intentó en la misma época, fracasando también, pero en esta ocasión la intoxicación le causó la muerte.5
Edmond Frémy (inicialmente ayudante de Louyel) abordó el tema con mucha mayor cautela y seguridad, lo que le valió librarse de la intoxicación. Regresó a la electrólisis y en el proceso fue el primero en obtener ácido fluorhídrico puro (anteriormente solo se lo obtenía mezclado con agua), pero tampoco logró el objetivo.5
El químico francés Henri Moissan, inicialmente ayudante de Frémy, continuó con el intento. Probó métodos químicos (usando fluoruro de fósforo) pero fracasó, por lo que decidió intentar con electrólisis. Usó fluoruro arsénico pero al comenzar a intoxicarse paso al ácido fluorhídrico, continuando la labor de su maestro. Para que condujera la electricidad agregó fluoruro de potasio al ácido fluorhídrico puro y logró la electrólisis.6 Para que el flúor no atacara al electrodo positivo, usó una aleación de platino e iridio, apoyado en fluorita como aislante, y adicionalmente realizó la electrólisis a 50 grados bajo cero.6 Finalmente, el 26 de junio de 1886, Moissan fue el primero que obtuvo flúor en forma pura, lo que le valió el Premio Nobel de Química de 1906.67
La primera producción comercial de flúor fue para la bomba atómica del Proyecto Manhattan, en la obtención de hexafluoruro de uranio, UF6, empleado para la separación de isótopos de uranio. Este proceso se sigue empleando para aplicaciones de energía nuclear.
Abundancia y obtención[editar]
El flúor es el halógeno más abundante en la corteza terrestre, con una concentración de 950 ppm. En el agua de mar esta se encuentra en una proporción de aproximadamente 1,3 ppm. Los minerales más importantes en los que está presente son la fluorita, CaF2, el fluorapatito, Ca5(PO4)3F y la criolita, Na3AlF6.
El flúor se obtiene mediante electrólisis de una mezcla de HF y KF. Se produce la oxidación de los fluoruros:
- 2F- → F2 + 2e-
En el cátodo se descarga hidrógeno, por lo que es necesario evitar que entren en contacto estos dos gases para que no haya riesgo de explosión
Compuestos[editar]

El oxígeno combustiona mejor con los HC porque siempre se forma CO2, en cambio con flúor pueden formarse perfluorcadenas que son bastante inertes. El compuesto más oxidante puede ser el O2)F2) o bien el ion XeF+. El flúor se puede obtener químicamente en reacciones de ácidos de Lewis.
- Se emplean numerosos compuestos orgánicos en los que se han sustituido formalmente átomos de hidrógeno por átomos de flúor. Hay distintas formas de obtenerlos, por ejemplo mediante reacciones de sustitución de otros halógenos: CHCl3 + 2HF → CHClF2 + 2HCl
- Los CFC se han empleado en una amplia variedad de aplicaciones, por ejemplo como refrigerantes, propelentes, agentes espumantes, aislantes, etc., pero debido a que contribuyen a la destrucción de la capa de ozono se han ido sustituyendo por otros compuestos químicos, como los HCF. Los HCFC también se emplean como sustitutos, pero también destruyen la capa de ozono, aunque en menor medida a largo plazo.
- El politetrafluoroetileno (PTFE), es un polímero denominado comúnmente teflón.
El fluoruro de hidrógeno es extremadamente corrosivo y reacciona violentamente con los alcalinos y el amoníaco anhidro. Destruye el tejido hasta el hueso, es más peligroso que el ácido sulfúrico y el ácido nítrico.
- El ácido fluorhídrico es una disolución de fluoruro de hidrógeno en agua. Es un ácido débil, pero mucho más peligroso que ácidos fuertes como el clorhídrico o el sulfúrico atraviesa la piel destruye los tejidos y huesos, y es tóxico en cualquier concentración, además provoca hipocalcemia. El HF anhidro es extraordinariamente corrosivo.
Las disoluciones de HF son mortales aunque sean diluidas.
- El hexafluoruro de uranio, UF6, es un gas a temperatura ambiente que se emplea para la separación de isótopos de uranio.
- El flúor forma compuestos con otros halógenos presentando el estado de oxidación -1, por ejemplo, IF7, BrF5, ClF, etcétera.
Dichos compuestos son muy reactivos: el ClF3 es aún más reactivo que el flúor, así como el BrF5,
- La criolita natural, Na3AlF6, es un mineral que contiene fluoruros. Se extraía en Groenlandia, pero ahora está prácticamente agotada, por lo que se obtiene sintéticamente para ser empleada en la obtención de aluminio.
El HF anhidro y el ácido nítrico mezclados disuelven a la mayoría de los metales de transición, incluido el tántalo.
Efectos biológicos[editar]

Aunque el flúor es demasiado reactivo para tener alguna función biológica natural, se incorpora a compuestos con actividad biológica. Compuestos naturales organofluorados son raros, el ejemplo más notable es el fluoroacetato, que funciona como una defensa contra los herbívoros de plantas en al menos 40 plantas en Australia, Brasil y África.8 La enzima adenosil-fluoruro sintasa cataliza la formación de 5'- desoxi-5'-fluoroadenosina. El flúor no es un nutriente esencial, pero su uso tópico en la prevención de la caries dental es bien reconocida. El efecto es tópico (aplicación sobre la superficie del esmalte), aunque antes de 1981 se consideró principalmente sistémico (por ingestión).9 Su uso sistémico está actualmente desaconsejado por muchos autores.
Isótopos[editar]
El flúor tiene un único isótopo natural, el 19F. Este isótopo tiene un número cuántico de espín nuclear de 1/2 y se puede emplear en espectroscopia de resonancia magnética nuclear. Se suele emplear como compuesto de referencia el triclorofluorometano, CFCl3 o el trifluoroacético TFA.
El 18F es un isótopo artificial emisor de positrones (emisor β+), que puede obtenerse por medio de un ciclotrón a partir del 18O (bajo la forma química de H218O). El 18F, por su emisión radiactiva (positrones, que al aniquilarse con los electrones del medio producen dos rayos gamma de 511 keV), se utiliza en el diagnóstico por tomografía por emisión de positrones (PET, de sus siglas en inglés), la cual tiene aplicaciones en Oncología, Neurología y Cardiología. El 18F se incorpora a moléculas orgánicas (proceso denominado "marcación con 18F"). Las mismas son aplicadas al paciente por medio de inyectables y el patrón de su distribución en el organismo permite el diagnóstico de tumores, zonas de baja perfusión cardíaca o cerebral, entre otras.
Precauciones[editar]
El flúor y el HF deben ser manejados con gran cuidado y se debe evitar totalmente cualquier contacto con la piel o con los ojos. El HF anhidro hierve a 19 °C, sus vapores son muy irritantes y tóxicos, sus descubridores murieron por su acción. Nunca ha de mezclarse con metales alcalinos ni con amoniaco. En presencia de SbF5, se convierte en un superácido (el HF anhidro). La capacidad de protonación es tan grande que oxida a metales como el cobre y protona al metano etc. Tanto el flúor como los iones fluoruro son altamente tóxicos. El flúor presenta un característico olor acre y es detectable en unas concentraciones tan bajas como 0,02 ppm, por debajo de los límites de exposición recomendados en el trabajo.
Toxicidad[editar]
La toxicidad del flúor viene por su afinidad a unirse al zinc (básico para el aprendizaje, la memoria y la formación de anticuerpos), y al yodo (básico para la tiroides y el sistema hormonal del cuerpo y otras funciones, siendo además el yodo quelante de mercurio), esto es similar al mercurio que se amalgama con el yodo y el zinc. Además, el exceso de flúor puede producir malformaciones óseas, aparte de un "endurecimiento y fragilidad" de los huesos con una mayor facilidad a su rotura. En definitiva, el flúor puede dañar el sistema de aprendizaje, memoria, salud, sistema hormonal, huesos, y así de energía y productividad de las personas.
En la característica de unión con el yodo, se usa el flúor para tratar hipertiroidismos (un hiperdesarrollo de la tiroides, entre otras cosas por exceso de yodo). Al eliminar el yodo del cuerpo, el flúor reduce la tiroides, reduciendo su tamaño y actividad, siendo esto muy dañino para personas con tiroides normales (que hace una parte vital del sistema hormonal del cuerpo), y especialmente para las personas con una tiroides débil o hipotiroidismo. Esto se ve agravado si la persona está expuesta a contaminación por mercurio (amalgama de los dientes 55 % mercurio, lámparas halógenas/fluorescentes-cuando se funden o parpadean, contaminación minera, pescado contaminado, aire contaminado con altos niveles de diésel y del mercurio expulsado por su combustión,10 etc.), pues el mercurio también se une al zinc y al yodo, inutilizando sus funciones, se refuerza en el daño con el flúor.
Un síntoma de intoxicación por flúor fácilmente perceptible en la población infantil (pues sus dientes están en formación), es la presencia de manchas blancas en los dientes.
A nivel histórico, como anécdotas, en los primeros experimentos de refinamiento de uranio para hacer la bomba atómica, se pensaba que toda la toxicidad del proceso venía del uso de flúor.
CLORO
El cloro es un elemento químico de número atómico 17 situado en el grupo de los halógenos (grupo VIIA) de la tabla periódica de los elementos. Su símbolo es Cl. En condiciones normales y en estado puro forma dicloro: un gas tóxicoamarillo-verdoso formado por moléculas diatómicas (Cl2) unas 2,5 veces más pesado que el aire, de olor desagradable y tóxico. Es un elemento abundante en la naturaleza y se trata de un elemento químico esencial para muchas formas de vida.

Características principales[editar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_Diego_Mateo_Zapata.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_(cropped).jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
En la naturaleza no se encuentra en estado puro ya que reacciona con rapidez con muchos elementos y compuestos químicos, por esta razón se encuentra formando parte de cloruros (especialmente en forma de cloruro de sodio), cloritos y cloratos , en las minas de sal y disuelto en el agua de mar.
Historia[editar]
El cloro (del griego χλωρος, que significa "verde pálido") fue descubierto en su forma diatómica en 1774 por el sueco Carl Wilhelm Scheele, aunque creía que se trataba de un compuesto que contenía oxígeno. Lo obtuvo a partir de la siguiente reacción:
- 2 NaCl + 2H2SO4 + MnO2 → Na2SO4 + MnSO4 + 2 H2O + Cl2
En 1810 el químico inglés Humphry Davy demuestra que se trata de un elemento físico y le da el nombre de cloro debido a su color. El gas cloro se empleó en la Primera Guerra Mundial, siendo el primer caso de uso de armas químicas como el fosgeno y el gas mostaza.
Abundancia[editar]
El cloro se encuentra en la naturaleza combinado con otros elementos formando principalmente sales iónicas; como es el caso del cloruro sódico y cálcico; también con la mayoría de metales; desde el cloruro de hafnio hasta el cloruro de plata. Podría decirse que el cloro combina de forma natural bastante bien con la mayoría de elementos, excepto con los de su grupo, halógenos y gases nobles, aunque en las últimas décadas de manera sintética forma parte de los mismos en compuestos conocidos como son los fluorocloruros y cloruros de xenón.
Finalmente cabe destacar que la gran mayoría de estos compuestos suelen encontrarse con impurezas formando parte de minerales como la carnalita, KMgCl3·6H2O.
Obtención[editar]
El cloro comercial se obtiene por electrólisis en el proceso de preparación de los álcalis y se expande en forma líquida, no es puro; y por lo tanto, ha de purificarse.
Si se trata el dióxido de manganeso hidratado con ácido clorhídrico concentrado se produce un gas exento en gran parte de impurezas tales como el oxígeno gas (O2(g)) y óxidos de cloro.
4HCl + MnO2xH2O = MnCl2 + (x+2)H2O + Cl2
Compuestos[editar]
- Algunos cloruros metálicos se emplean como catalizadores. Por ejemplo, FeCl2, FeCl3, AlCl3.
- Ácido clorhídrico, HCl. Se emplea en la industria alimentaria, metalúrgia, desincrustante, productos de limpieza, abrillantador de pisos, destapador de caños y tuberías.
- Ácido hipocloroso, HClO. Se emplea en la depuración de aguas y alguna de sus sales como agente blanqueante.
- Ácido cloroso, HClO2. La sal de sodio correspondiente, NaClO2, se emplea para producir dióxido de cloro, ClO2, el cual se usa como desinfectante.
- Ácido clórico (HClO3). El clorato de sodio, NaClO3, también se puede emplear para producir dióxido de cloro, empleado en el blanqueo de papel, así como para obtener clorato.
- Ácido perclórico (HClO4). Es un ácido oxidante y se emplea en la industria de explosivos. El perclorato de sodio, NaClO4, se emplea como oxidante y en la industria textil y papelera.
- Compuestos de cloro como los clorofluorocarburos (CFC) contribuyen a la destrucción de la capa de ozono.
- Algunos compuestos orgánicos de cloro se emplean como pesticidas. Por ejemplo, el hexaclorobenceno (HCB), el para-diclorodifeniltricloroetano (DDT), el toxafeno, etcétera.
- Muchos compuestos organoclorados presentan problemas ambientales debido a su toxicidad, por ejemplo el pentacloroetano, los pesticidas anteriores, los bifenilos policlorados (PCB), o las dioxinas.
Isótopos[editar]
En la naturaleza se encuentran dos isótopos estables de cloro. Uno de masa 35 uma, y el otro de 37 uma, con unas proporciones relativas de 3:1 respectivamente, lo que da un peso atómico para el cloro de 35,5 uma.
El cloro tiene 9 isótopos con masas desde 32 uma hasta 40 uma. Solo tres de éstos se encuentran en la naturaleza: el 35Cl, estable y con una abundancia del 75,77 %, el 37Cl, también estable y con una abundancia del 24,23 %, y el isótopo radiactivo 36Cl. La relación de 36Cl con el Cl estable en el ambiente es de aproximadamente 700 × 10−15:1.
El 36Cl se produce en la atmósfera a partir del 36Ar por interacciones con protones de rayos cósmicos. En el subsuelo se genera 36Cl principalmente mediante procesos de captura de neutrones del 35Cl, o por captura de muones del 40Ca. El 36Cl decae a 36S y a 36Ar, con un periodo de semidesintegración combinado de 308000 años.
El período de semidesintegración de este isótopo hidrofílico y no reactivo lo hace útil para la datación geológica en el rango de 60 000 a un millón de años. Además, se produjeron grandes cantidades de 36Cl por la irradiación de agua de mar durante las detonaciones atmosféricas de armas nucleares entre 1952 y 1958. El tiempo de residencia del 36Cl en la atmósfera es de aproximadamente una semana. Así pues, es un marcador para las aguas superficiales y subterráneas de los años 1950, y también es útil para la datación de aguas que tengan menos de 50 años. El 36Cl se ha empleado en otras áreas de las ciencias geológicas, incluyendo la datación de hielo y sedimentos.
| Núclido | Abundancia | Masa | Espín | Periodo de semidesintegración | Producto de desintegración |
| 32Cl | - | 31,9857 | 1 | 298 ms | ε |
| 33Cl | - | 32,9775 | 3/2 | 2,51 s | ε |
| 34Cl | - | 33,9738 | 0 | 1,53 s | ε |
| 35Cl | 75,77 | 34,9689 | 3/2 | - | - |
| 36Cl | - | 35,9683 | 2 | 301000 a | β- |
| 37Cl | 24,23 | 36,9659 | 3/2 | - | - |
| 38Cl | - | 37,9680 | 2 | 37,2 m | β- |
| 39Cl | - | 38,9680 | 3/2 | 55,6 m | β- |
| 40Cl | - | 39,9704 | 2 | 1,38 m | β- |
| 41Cl | - | 40,9707 | n.m. | 34 s | β- |
| 42Cl | - | 41,9732 | n.m. | 6,8 s | β- |
| 43Cl | - | 42,9742 | n.m. | 3,3 s | β- |
Aplicaciones y usos[editar]
Producción de insumos industriales y para consumo[editar]
Las principales aplicaciones de cloro son en la producción de un amplio rango de productos industriales y para consumo.12 Por ejemplo, es utilizado en la elaboración de plásticos, solventes para lavado en seco y desgrasado de metales, producción de agroquímicos y fármacos, insecticidas, colorantes y tintes, etc.
Purificación y desinfección[editar]
El cloro es un químico importante para la purificación del agua (como en plantas de tratamiento de agua), en desinfectantes, y en la lejía. El cloro en agua es más de tres veces más efectivo como agente desinfectante contra Escherichia coli que una concentración equivalente de bromo, y más de seis veces más efectiva que una concentración equivalente de yodo.3
El cloro como antiséptico fue introducido en 1835 por Holmes (en Boston) y 1847 Semmelweis (en Viena).4 El cloro se emplea como desinfectante en mobiliarios, equipos, instrumental y áreas hospitalarias.4 El cloro suele ser usado en la forma de ácido hipocloroso para eliminar bacterias, hongos, parásitos y virus en los suministros de agua potable y piscinas públicas. En la mayoría de piscinas privadas, el cloro en sí no se usa, sino hipoclorito de sodio, formado a partir de cloro e hidróxido de sodio, o tabletas sólidas de isocianuratos clorados. Incluso los pequeños suministros de agua son clorados rutinariamente ahora.5 (Ver tambiéncloración)
Suele ser impráctico almacenar y usar el venenoso gas cloro para el tratamiento de agua, así que se usan métodos alternativos para agregar cloro. Estos incluyen soluciones de hipoclorito, que liberan gradualmente cloro al agua, y compuestos como la dicloro-S-triazinatriona de sodio (dihidrato o anhidro), algunas veces referido como "diclor", y la tricloro-S-triazinatriona, algunas veces referida como "triclor". Estos compuestos son estables en estado sólido, y pueden ser usados en forma de polvo, granular, o tableta. Cuando se agrega en pequeñas cantidades a agua de piscina o sistemas de agua industrial, los átomos de cloro son hidrolizados del resto de la molécula, formando ácido hipocloroso (HClO), que actúa como un biocida general, matando gérmenes, microorganismos, algas, entre otros de ahí su importancia en el empleo en Endodoncia como agente irrigante de los conductos radiculares abordándose como solución en forma de hipoclorito de sodio en distintas concentraciones sea 0,5 % o 0,2 % las más frecuentes empleadas. El cloro también es usado como detergente para bacterias como el Bacillus repridentius o como el Martelianus marticus.

GRUPOS BUEN TRABAJO 5,O
ResponderEliminarJUEGO 1,O
FINAL 3,0
El juego no se distinguen las imágenes
ResponderEliminardefinitiva nivelación 4.2